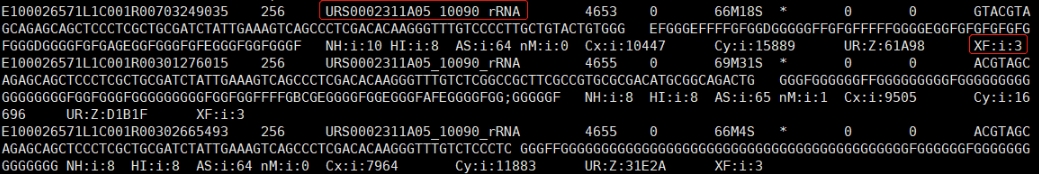
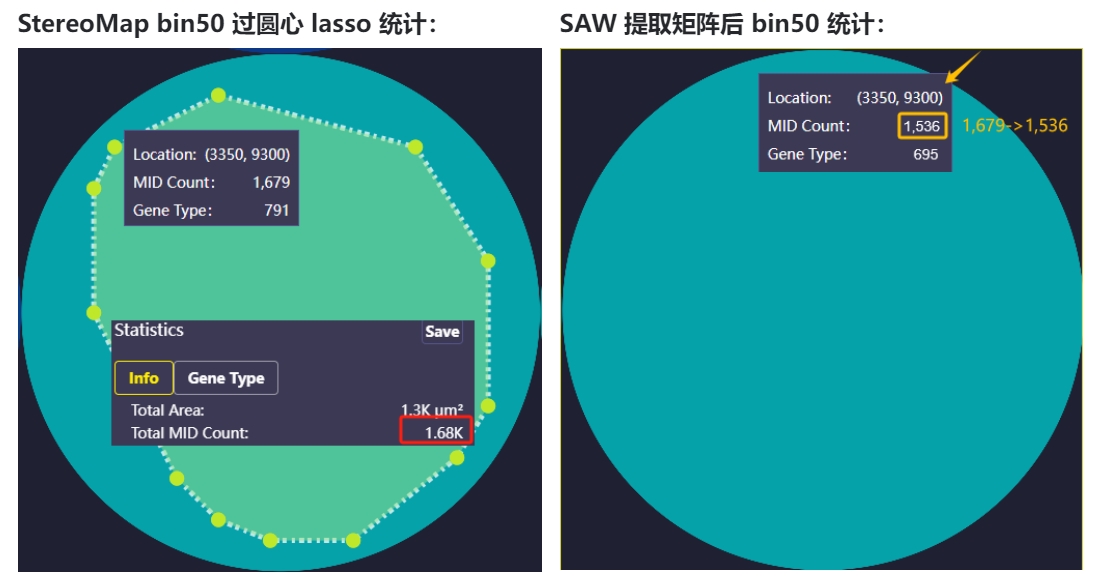
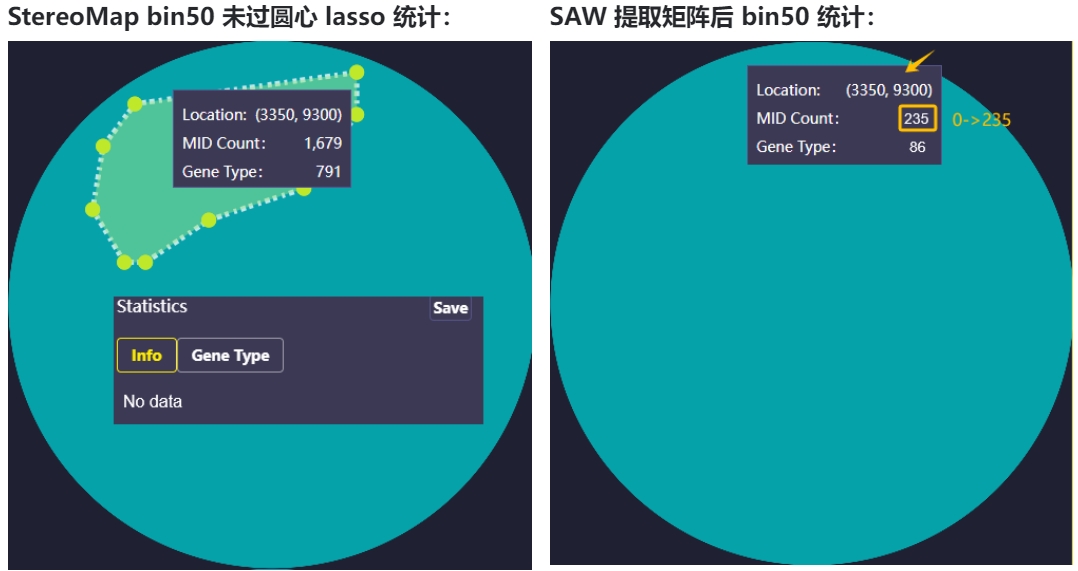
1、方式一 (SAW < v8.0.0)
①可以使用SAW sif中的checkGTF工具进行检查,运行方式如下:
## export SINGULARITY_BIND="/path/to/input/dir,/path/to/output/dir"
singularity exec SAW.sif checkGTF \
-i <input.gtf/gff> \ ## GTF/GFF file input to be checked
-o <output.gtf/gff> ## [optional]. Set to output revised GTF/GFF file. Be aware that this may remove some genes which do not meet the requirements and cannot be fixed
②可能有部分无法被程序修复的基因注释记录会在输出时被删除,可以在日志中找到记录,重新修改GTF文件后再次尝试。
2、方式二 (SAW >= v8.0.0)
①可以使用SAW checkGTF分析流程进行检查,运行方式如下:
SAW checkGTF \
-input-gtf=/path/to/input/GTF/or/GFF \
-output-gtf=/path/to/output/GTF/or/GFF
②可能有部分无法被程序修复的基因注释记录会在输出时被删除,可以在日志中找到记录,重新修改GTF文件后再次尝试。