
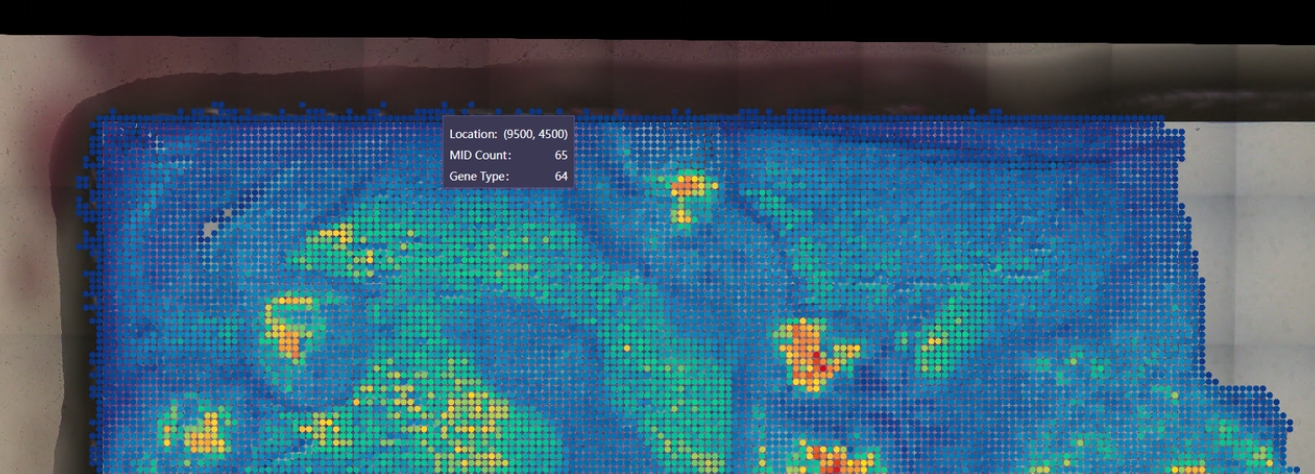
这种情况是正常的,以上图为例,图为图像+矩阵热图的可视化展示,原因主要有两点:一,由于样本切片位于芯片边缘,图像组织分割结果会生成在矩阵外周,造成“外溢”的假象;二,在观察矩阵热图时通常选择bin20或者bin50(将其视为一个spot单位点)的bin size尺寸,bin的坐标位点计算是取其左上角顶点,当实际表达只占spot单位点的一小部分时,可视化展示也会将整个spot点亮,所以矩阵边缘看起来像是“外扩”。



这种情况是正常的,以上图为例,图为图像+矩阵热图的可视化展示,原因主要有两点:一,由于样本切片位于芯片边缘,图像组织分割结果会生成在矩阵外周,造成“外溢”的假象;二,在观察矩阵热图时通常选择bin20或者bin50(将其视为一个spot单位点)的bin size尺寸,bin的坐标位点计算是取其左上角顶点,当实际表达只占spot单位点的一小部分时,可视化展示也会将整个spot点亮,所以矩阵边缘看起来像是“外扩”。
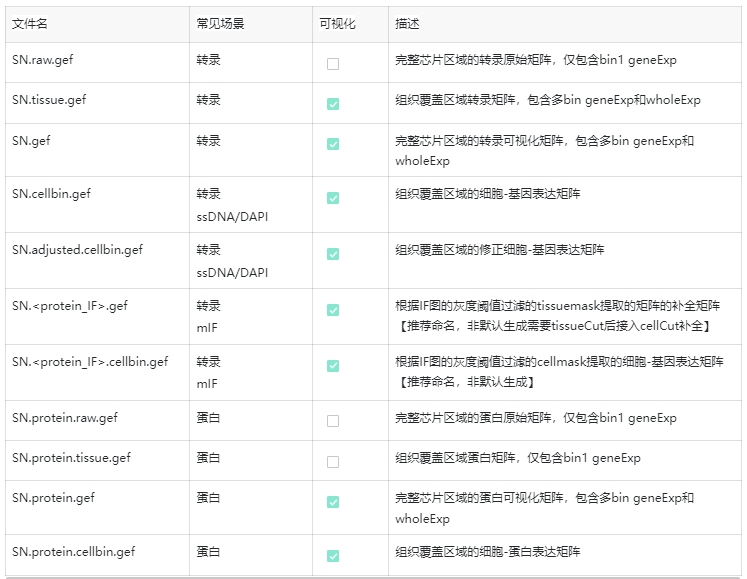
使用 SAW >= v8.0.0 运行SAW count分析流程时,如遇到因集群意外停机、环境kill任务或者队列爆内存情况,在重启集群/服务器后,重新提交任务,程序可以rerun分析,但是要求两次分析任务的命令参数完全一致,不可有任何更改,程序会对此进行校验,如有区别,会当成第二次不同的任务启动。
注:此功能不适用于参数设置错误,或者输入数据错误等人为因素引发的问题。
SAW分析流程入参时支持Q40的FASTQ和Q4的FASTQ文件。
Q40:以41种质量值描述所有的测序完成的碱基质量的质量体系。Q40 FASTQ为常见的PE形式、成对出现的数据,通常命名为<slide>_<lane_number>_<read_index>.fq.gz,包含read 1 和 read 2,少数情况下可能没有"read",此时可以检查是否为成对出现,或者检查文件格式。
## Q40 read 1
@V350156489L4C001R00100001484/1
TCTGCAGCCAACATGGACAGATCCTTTTAGAACTT
+
D>DC<CBEADEABCB(AADCDD2DD"DBE*$F'C'
## Q40 read 2
@V350156489L4C001R00100001484/2
CTATGAAACACACTATCCTCAATCGGCTCCTTAATTTCAATACCAGCCGT
+
ECFCCB?CAECEBDBCCFDFEFDF?<FCF>B?2EC=C?C<CE(AA=;B5B
Q4:以4种质量值描述所有的测序完成的碱基质量的质量体系。Q4 FASTQ为一组16或64个单个文件出现,通常命名为<slide>_<lane_number>_<sample_barcode>_<split_index>.fq.gz。少数情况下可能文件前缀中没有barcode,通过文件目录中的barcode目录名区分。Q4 FASTQ经常也会被叫做SE FASTQ,其实实际依然是双端测序,但在完成测序下机写出FASTQ时,以单个文件的方式输出,看起来和SE数据一样。这种格式将read1中的信息编码后写入read 2的readID中,同时序列质量信息以Q4(以4种质量值描述所有的测序完成的碱基质量的质量体系)方式记录,以降低存储。

## Q4 read
@FP300000513L1C002R00400000218 CE242DF29A57 97D26
GTGTAGTGAACCCCATGGTAGTTTTCTGATTGTTGTTAAAAAAAATGACTTAACATATTACATGGACACTCAATAAAAATGTTTTATTTCCTGTTGAAAA
+
FFFFFFFFFFFF8F8FFFFFFFFFFFFF8FFFFFFFFF8FF8FFF8FFFFFFF,FFFFFFFFFFF8FFFFFF8F8F,F8FFFFFF,FFFFFFFFFF,FFF
可通过设置--genomeChrBinNbits
=min(18,log2[max(GenomeLength/NumberOfReferences,ReadLength)])来降低RAM消耗,例如:14G大小的参考基因组,其chr/scaffolds为90k,根据上述公式,计算发现--genomeChrBinNbits的参数值应设置为17。详情可参考STAR软件的说明手册https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf。
注:SAW 软件版本 < 8.0
1、可以邻片H&E切片圈组织结构,受临片配准精度限制,组织分割结果需要用户手动介入进行调整;
2、H&E染色做细胞分割,时空StereoMap软件兼容任何第三方软件的细胞分割结果。
lncRNA MID/Total MID:人样本为6%~12%;小鼠样本为5%~9%(该数据针对部分研发数据分析,因受样本量限制,您的实测数据中该比例可能会略有差异)。
在StereoMap软件上直接lasso感兴趣区域即可,StereoMap操作流程可参考时空官网,链接为:https://www.stomics.tech/products/BioinfoTools/OfflineSoftware。
1、对于微生物的检测种类,主要看数据库的选择。我们提供了相对权威的数据库,但用户也可以自己配置其他数据库到流程中;
2、目前对于微生物可以定位到“种”,目前是看不到捕获的微生物基因数。
FFPE转录组产品支持Cellbin,可通过以下两种方式实现:
① 基于ssDNA染色,可以实现Cellbin分割功能(目前主要小鼠样本);
② 时空StereoMap软件兼容任何第三方算法单细胞分割的结果。