
核染色图像
为什么要做核染色图像?
核染色在组织切片中不存在组织分离偏倚,对于确定组织区域和细胞位置具有较高的价值。Stereo-seq 实验和生信分析工具同时兼容了 ssDNA 和 DAPI 两种核染色试剂。
STOmics 的研发团队测试并对比了多种商业化的染色试剂,发现 ssDNA 染色对下游 mRNA 捕获率的影响最小,DAPI 对染色细胞核具有高度特异性,可以与其它荧光试剂一起使用,是一种常用的染料。这两种染色方法都可以看到轨迹线。
核染色图像的注意事项
StereoMap 和 SAW 中处理的核染色图像为单通道的 8 位/ 16 位深的灰度图像,或者是 RGB 彩色图像。 (有关更多信息,请参见 图像类型和格式 获取更多信息。)
| 荧光图像 | 数据类型和格式 |
|---|---|
| 灰度图像 | 8 / 16 位的单通道图像 |
第一步:上传图像
.png)
点击选择框中的 Choose file,选择一个兼容的图像文件或者也可以将所选文件直接拖入到选择框中,核染色图像类型仅允许上传一个图像文件。
请参考图像处理输入文件获取更多的图像文件信息。
.png)
选择文件后会触发一个文件解析的过程,该过程不仅会读取图像,还会从输入中获取必要的信息,解析的时间会因文件类型和图像大小有所差异。在图像处理步骤,Stereo-seq 芯片序列号(SN)和显微镜配置会提供重要的参考信息,如果输入的文件是.tar.gz 或者 .stereo,信息已写入到输入文件中,可以直接开始解析。
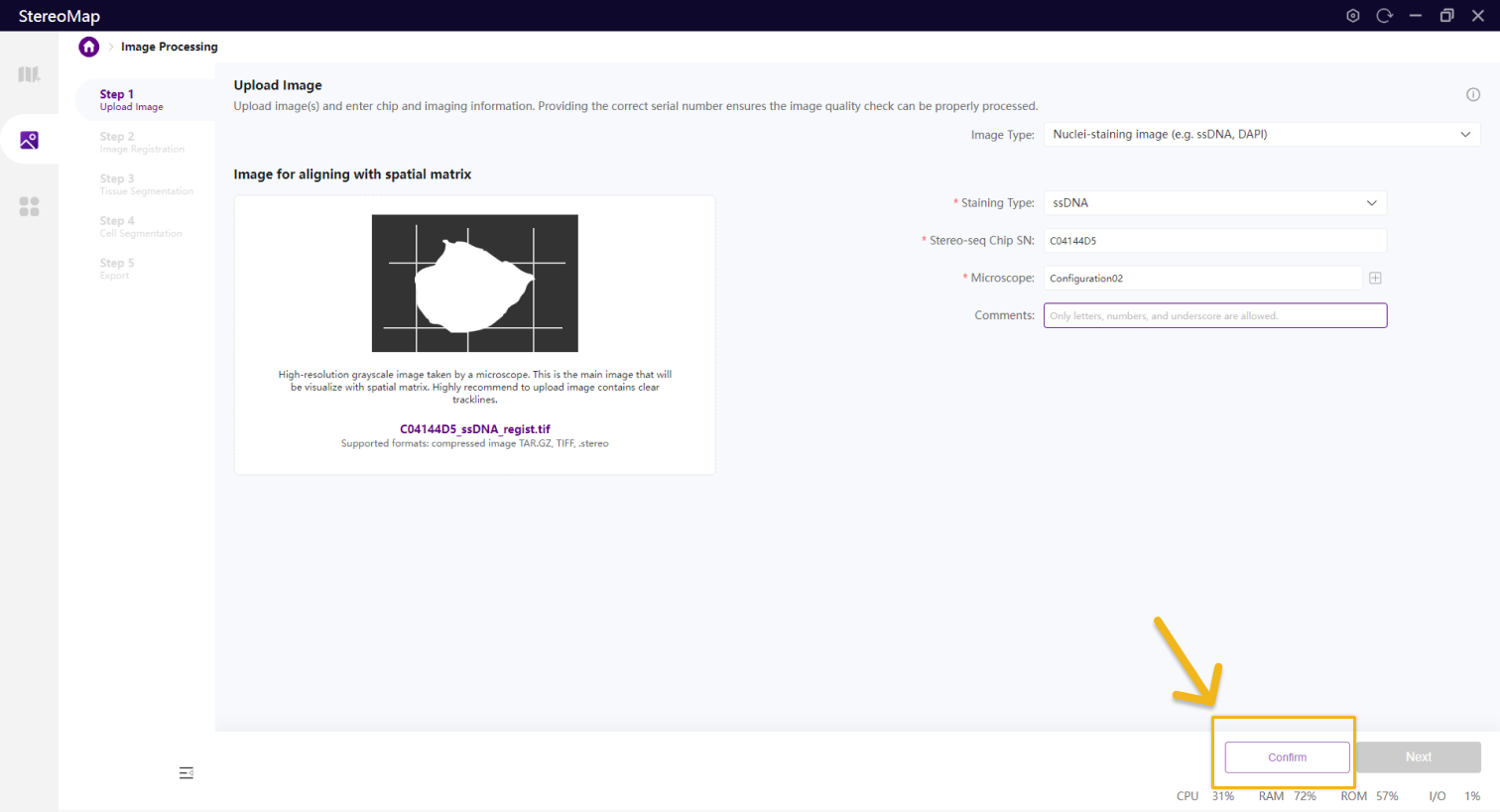
如果输入的文件格式为.tif 或者.tiff,则需要用户输入指定的必要信息并点击 Confirm 按钮后才可以做图像的解析和质控分析,一般情况下,您可以选择显微镜配置来输入有关显微镜的信息,请参阅显微镜信息配置获取更多信息。
第二步:图像配准
在该步骤中,您需要调整图像的方向、角度和尺度,以使其与空间特征表达矩阵对齐。StereoMap 提供了两种配准方法,用于在 Stereo-seq 数据分析中对齐图像。
- 形态学配准:根据组织形态的相似度将图像对齐。
- 打点配准:通过标记特定的 track 点,将图像与从 Stereo-seq 芯片 SN 推导出来的 track 线模板对齐。该方法无需 SAW 生成的空间特征表达矩阵,但应考虑一些先决条件。
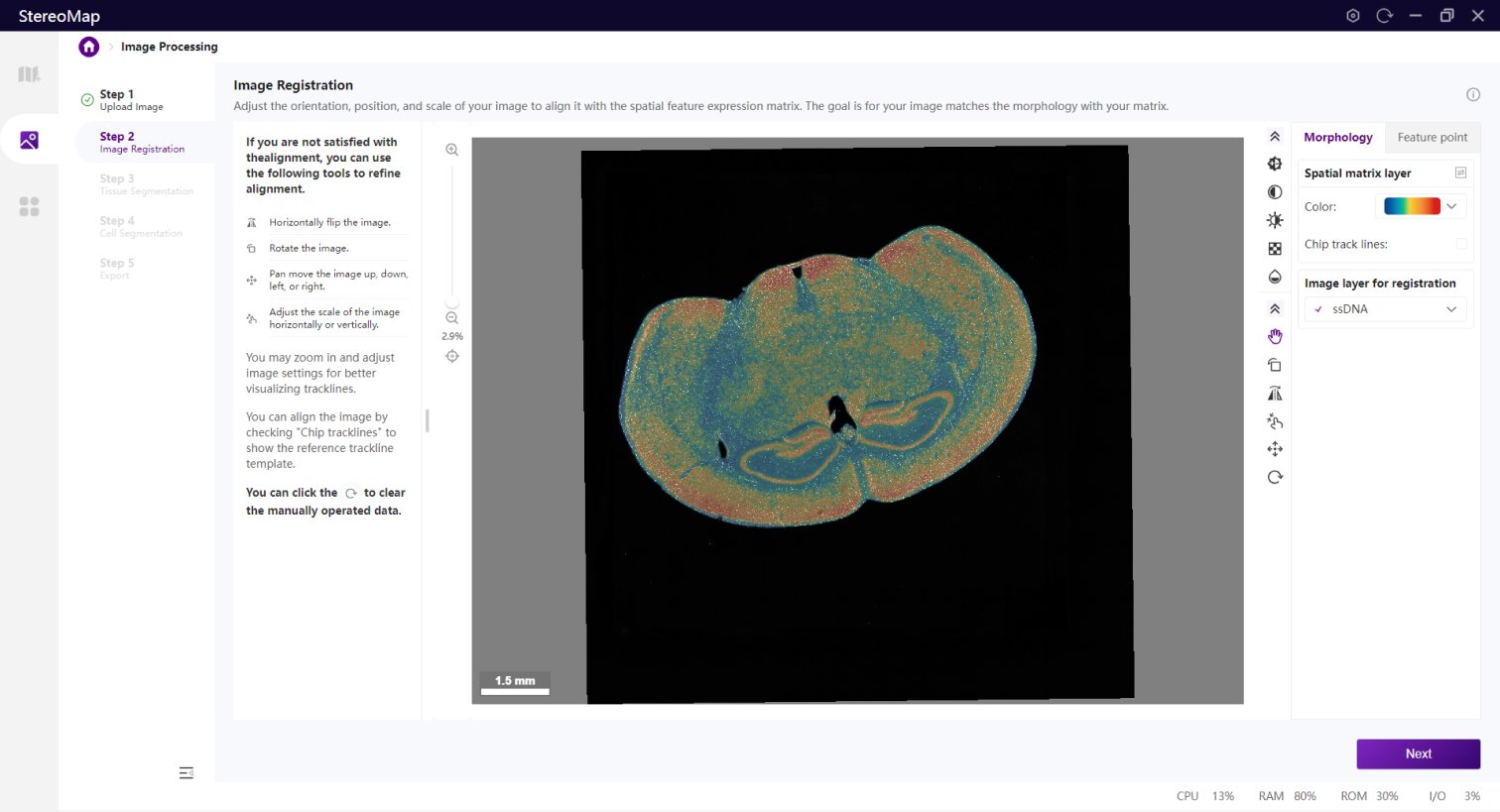
形态学配准
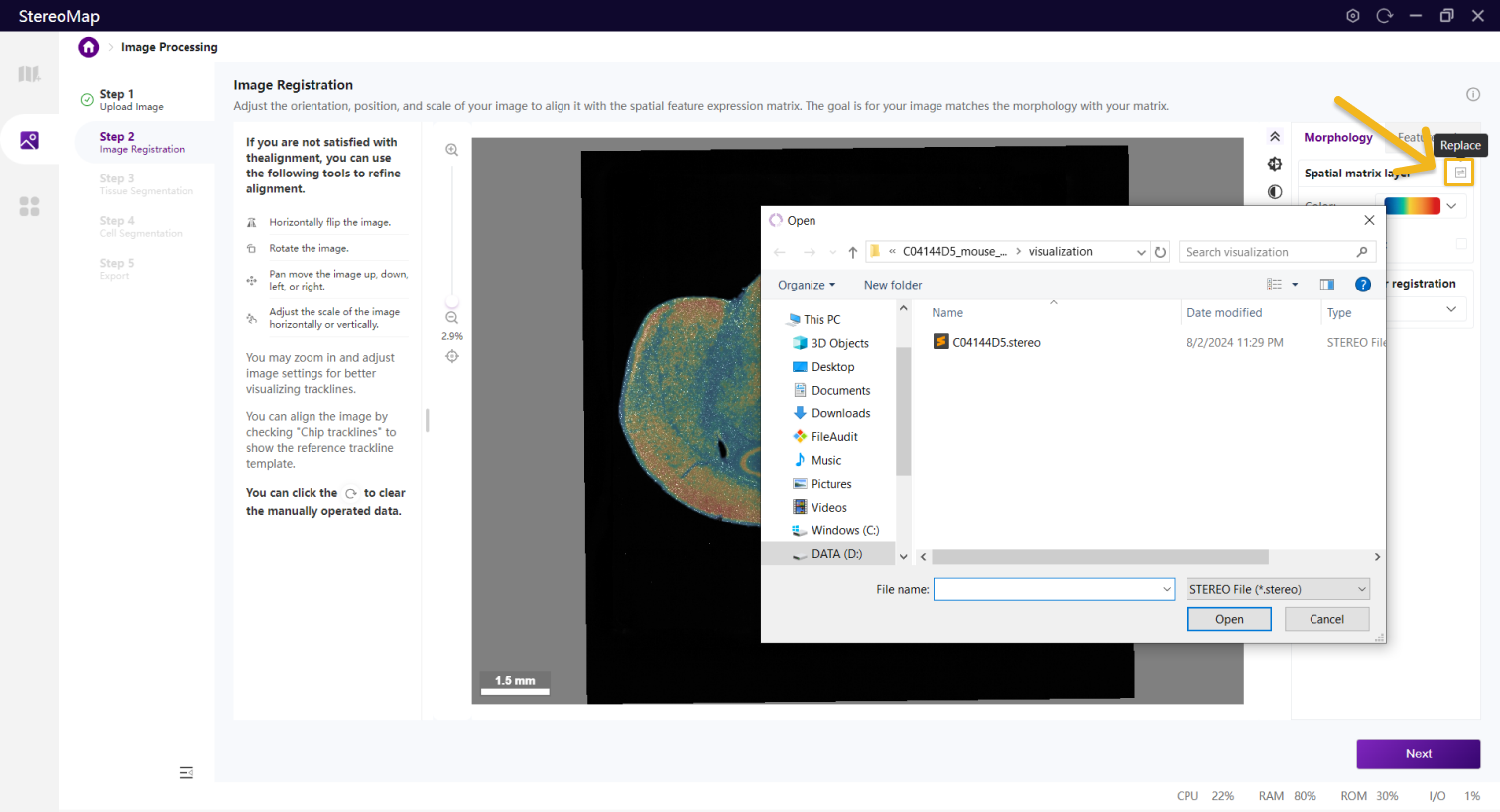
如果您在第 一步中:上传了 .stereo文件,进入第二步时,您就可以同时看到图像和空间特征表达矩阵;上传了 .tar.gz 或 .tif/.tiff 图像文件,在第二步您需要选择一个.stereo 文件来指定空间特征表达矩阵,可以点击  选择
选择 .stereo 文件。此外,如果您在第一步上传了.stereo 文件(包含图像文件),想更改第二步展示的空间特征表达矩阵文件,您可以点击 重新加载一个新的
重新加载一个新的.stereo 文件,但还是使用第一步上传的.stereo文件中的图像数据。
手动配准过程包括两个阶段:根据组织形态粗略的匹配图像的方向,然后精细调整图像的位置和尺度,确保与空间特征表达矩阵完全重叠。您还可以通过 Chip trackline,辅助进行精细对齐。
为了粗略对齐图像,您需要将显微镜图像与特征矩阵方向变换为一致。
- 使用翻转工具
 可以将图像绕Y轴镜像翻转。
可以将图像绕Y轴镜像翻转。 - 点击旋转按钮
 使用
使用 可以将图像以相同的方向旋转。
可以将图像以相同的方向旋转。
当图像方向跟表达矩阵方向一致时,您就可以继续进行精细对齐了。
做精细配准时,您需要将图像移动到有组织覆盖的区域。
- 通过平移面板
 设置移动的步长和移动的四个方向(上、下、左、右)。
设置移动的步长和移动的四个方向(上、下、左、右)。 - 因显微镜拍摄的图像数据的尺寸跟特征矩阵不同,您可以使用缩放工具
 来调整缩放的比例。
来调整缩放的比例。
您可以勾选 Chip trackline 显示参考轨迹线模板,辅助对齐图像。此时,参考轨迹线模板可以作为矩阵的一种表示形式,如果轨迹线较暗,可以手动调整图像的归一化 、对比度
、对比度 、亮度
、亮度 和不透明度
和不透明度 等工具按钮。
等工具按钮。
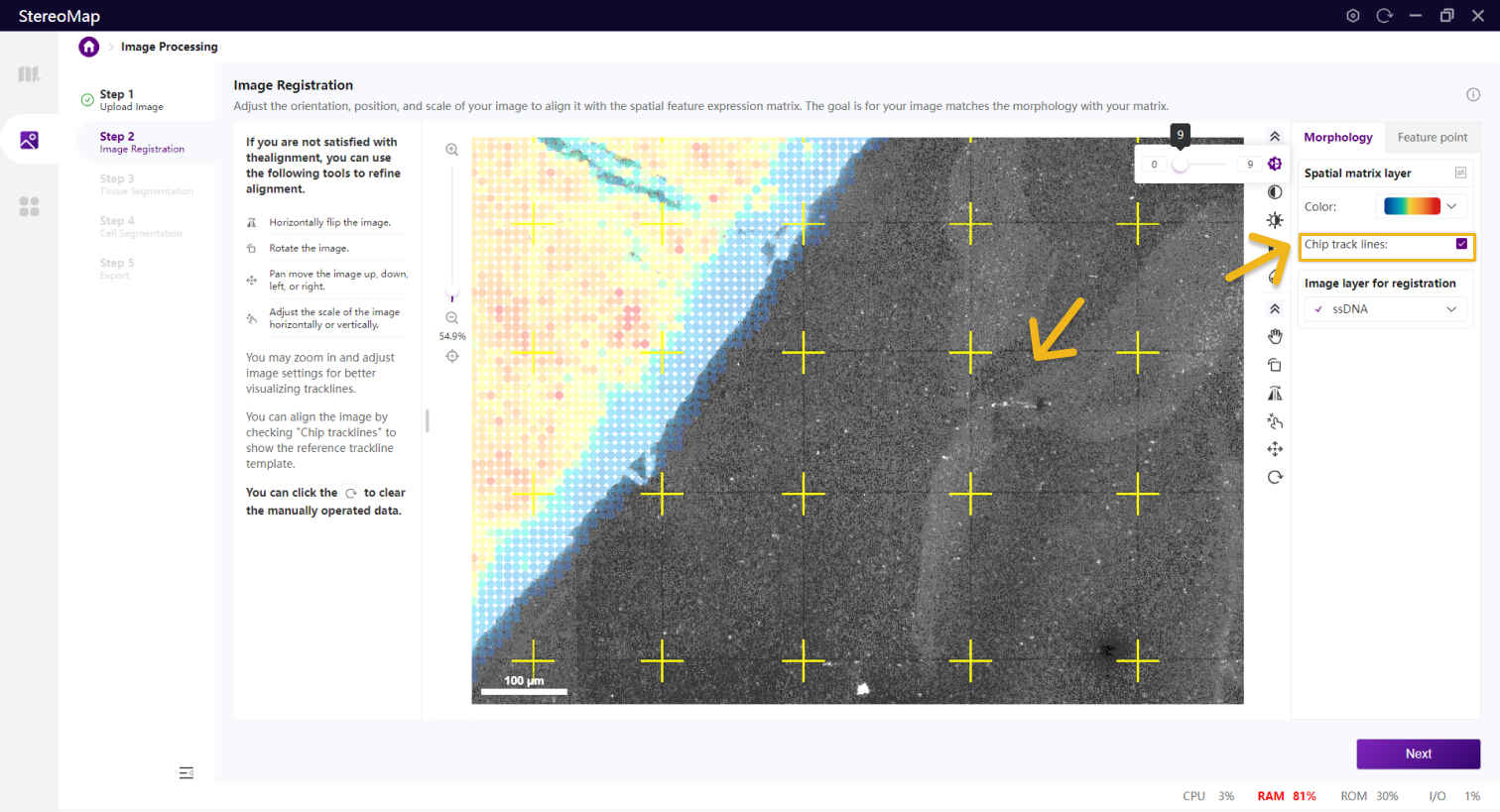
调整图像以便可以清楚的展示轨迹线。

饱和度 的调整对灰度图像不适用。
的调整对灰度图像不适用。
完成配准的图像将标记为已完成 。
。
如果做了手动配准,可以跳过步骤 3 和 4,您可以导出手动后的tar.gz图像文件,输入到 SAW 运行自动的组织分割和细胞分割。
另外,我们会输出*regist.tif 图像文件,该文件是配准后的图像,其形状和方向跟基因表达矩阵相匹配,此图像可以作为组织分割或者细胞分割的起点。如果您考虑使用第三方分割工具,强烈建议使用此图像文件作为输入,避免图像与 Mask 之间存在位移、角度和尺度的偏差。
打点配准
打点配准主要是在矩阵文件未生成的情况下,利用图像上的 track 点以及预定义规则的芯片 mask 信息将图像对齐。
您需要确认图像以选择到正确的用于计算的特征点,可能会导致您的数据配准失败。如果无法达到上述要求,强烈建议您使用形态学配准的方式做后续分析。
使用该方法的前提条件:
- 图像 QC 通过,因为要确保 track 线是可见的。
- 有预定义规则的 Stereo-seq 芯片: Stereo-seq N FFPE V1.0, Stereo-seq T FF V1.3或者Stereo-CITE T V1.1 的芯片。
- 输入
.tar.gz或.stereo文件中对应的.tar.gz是由 StereoMap >= 4.1 生成的。
为确保图像可以配准成功,需要您选择一个指定的点确定图像的方向,否则图像配准可能会失败。
选择特定的 track 点保证获取正确配准结果的要求如下:
如果您的图像数据无法满足以上所有的要求,强烈推荐您使用形态学配准。
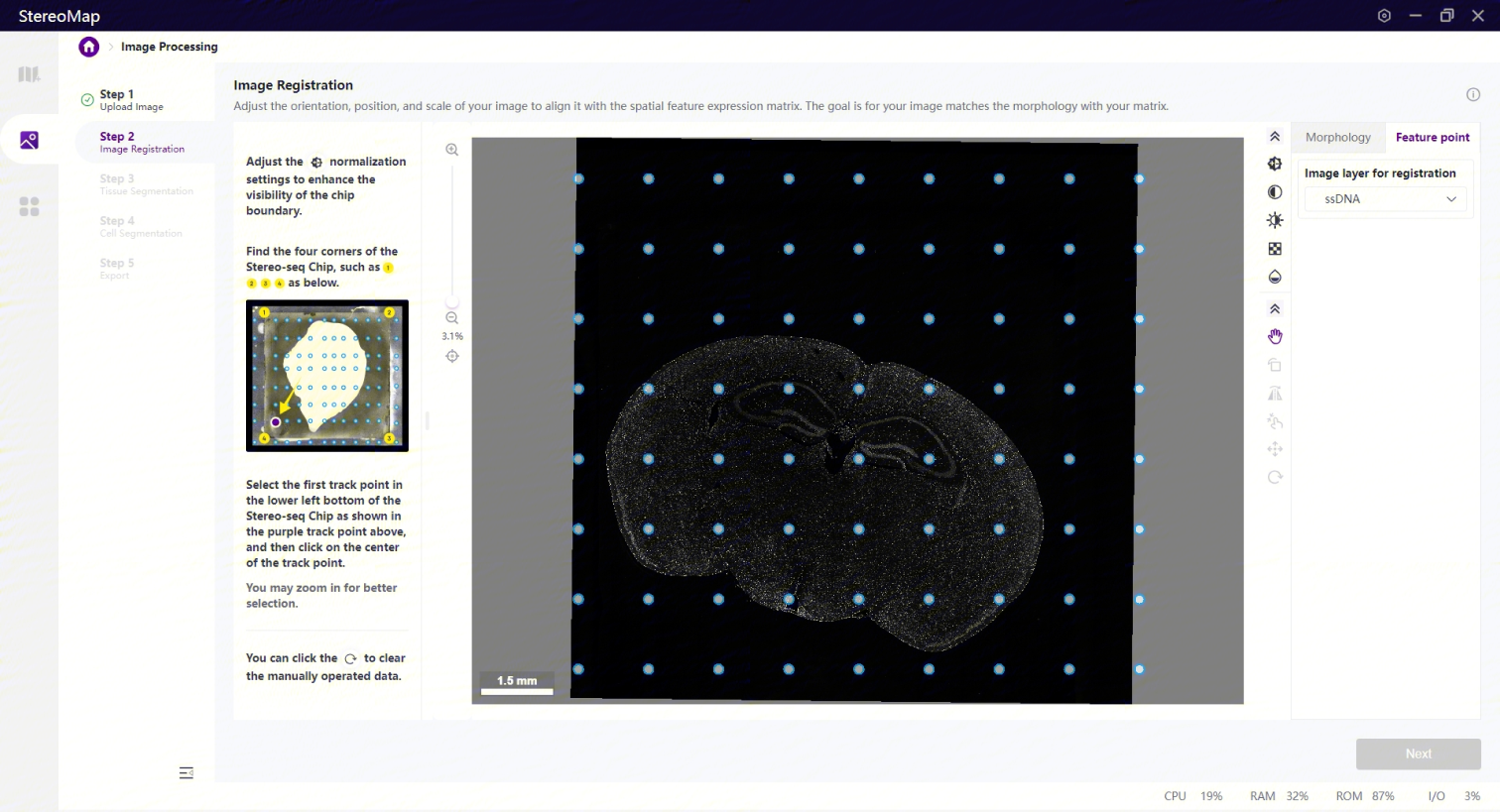
打点配准展示的图像数据为经过小角度旋转或者小尺度缩放的图像。
打点配准分为如下两个步骤:首先调整图像,确认芯片的四个边/角可以被清楚的看到。最后从已有的track点中选择某个track点将其作为参考点。
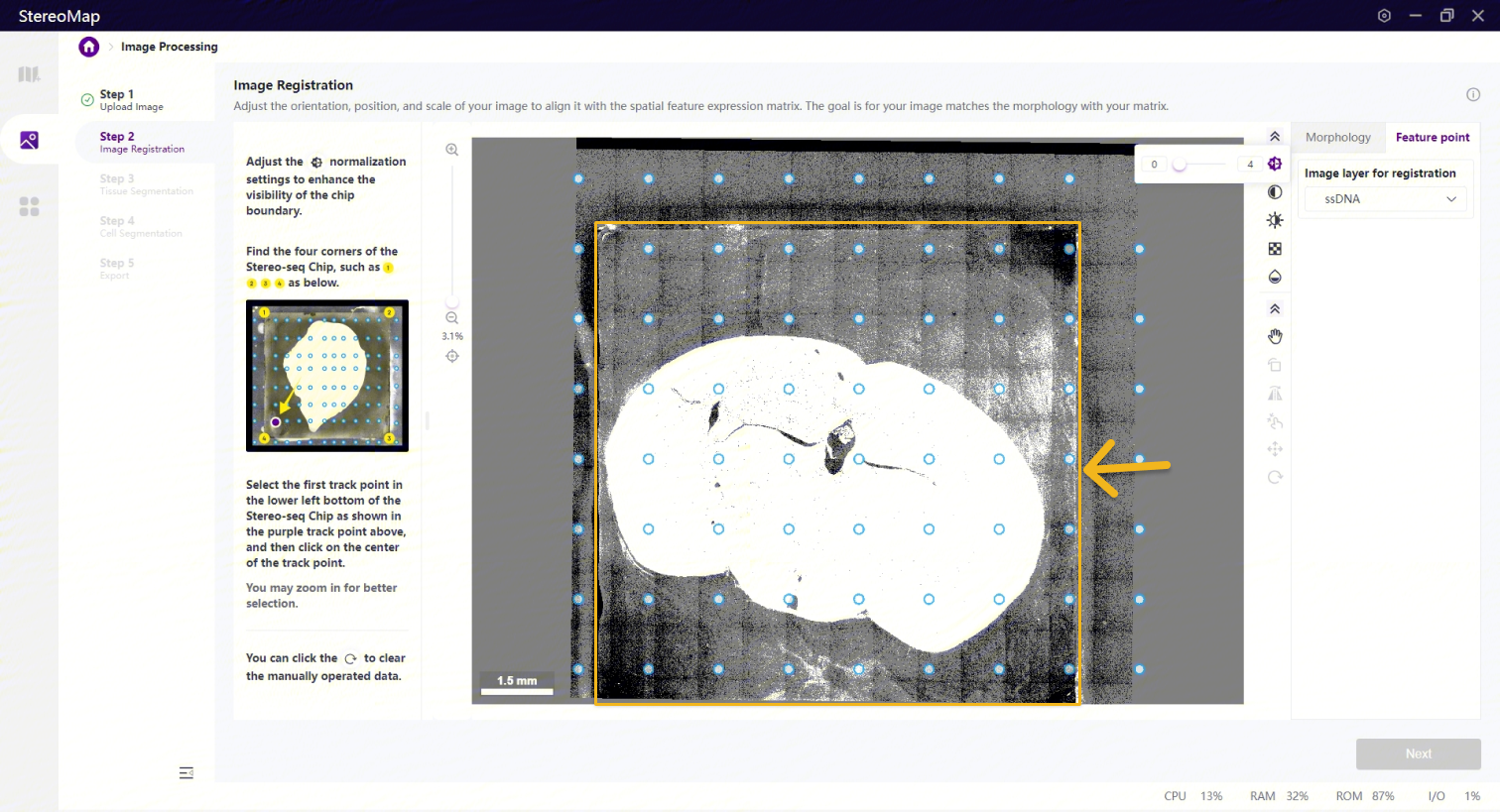
为保证图像的四个边/角可以被清晰的看到,您可以使用归一化、对比度、亮度和不透明度等工具调整图像。
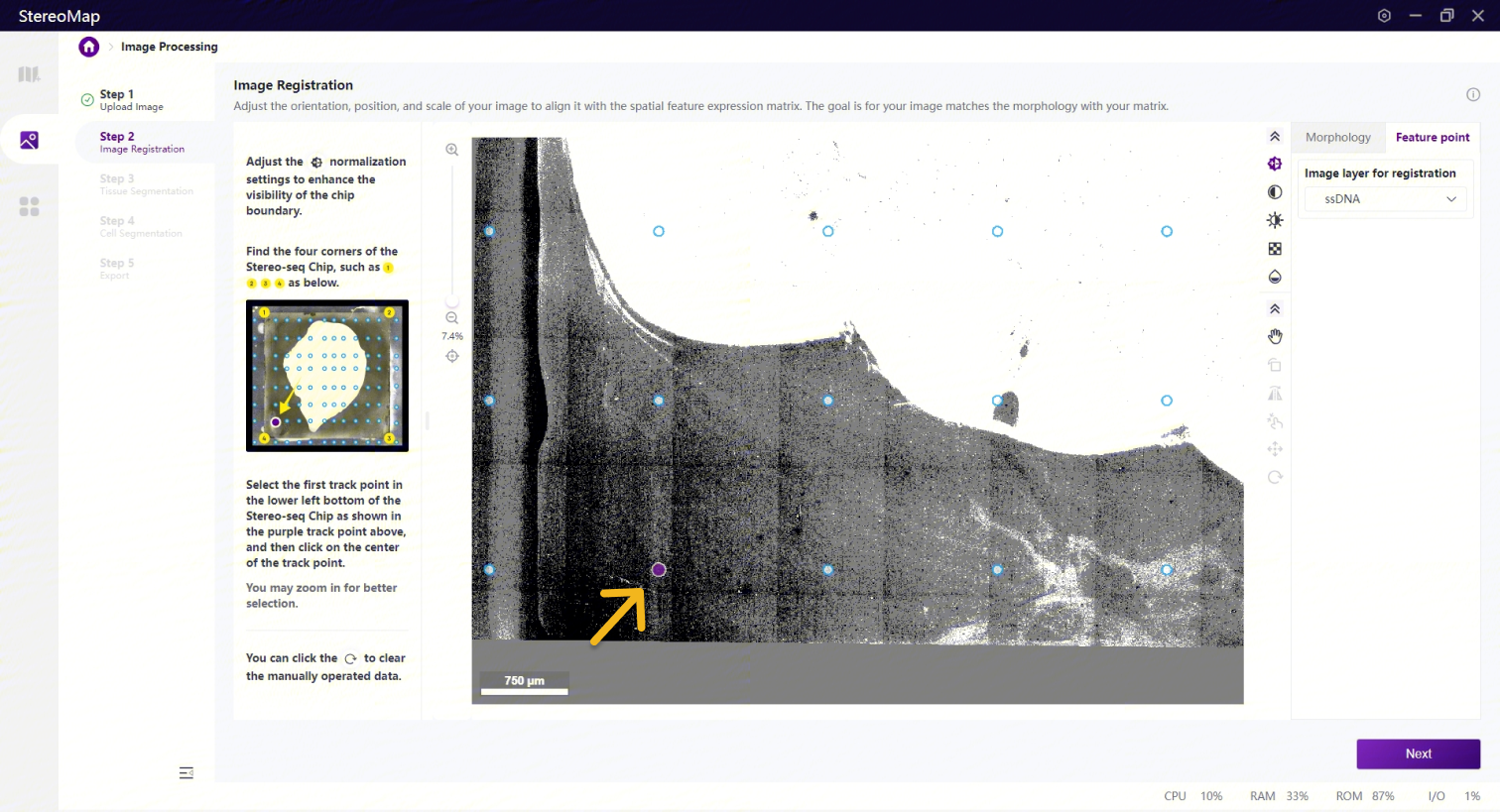
看到芯片的四个角后,您需要确认靠近芯片左下角的第一个预设的 track 点,单击该点选中后,点击 Next 按钮即可完成配准。

完成配准选特征点操作后,图像的方位将会被调整,并在 Step 3 组织分割步骤展示。
如果做了手动配准,可以跳过步骤 3 和 4,您可以导出手动后的tar.gz图像文件,输入到 SAW 运行自动的组织分割和细胞分割。
另外,我们会输出*regist.tif 图像文件,该文件是配准后的图像,其形状和方向跟基因表达矩阵相匹配,此图像可以作为组织分割或者细胞分割的起点。如果您考虑使用第三方分割工具,强烈建议使用此图像文件作为输入,避免图像与 Mask 之间存在位移、角度和尺度的偏差,导致第三方分割的Mask文件无法导入。
第三步:组织分割
组织分割是可跳过的步骤。
在该步骤中,您需要识别组织区域,准确的识别组织边界可以减少背景干扰对于聚类结果的影响。基于图像的组织分割结果将映射到空间特征表达矩阵上,生成组织区域的表达热图。
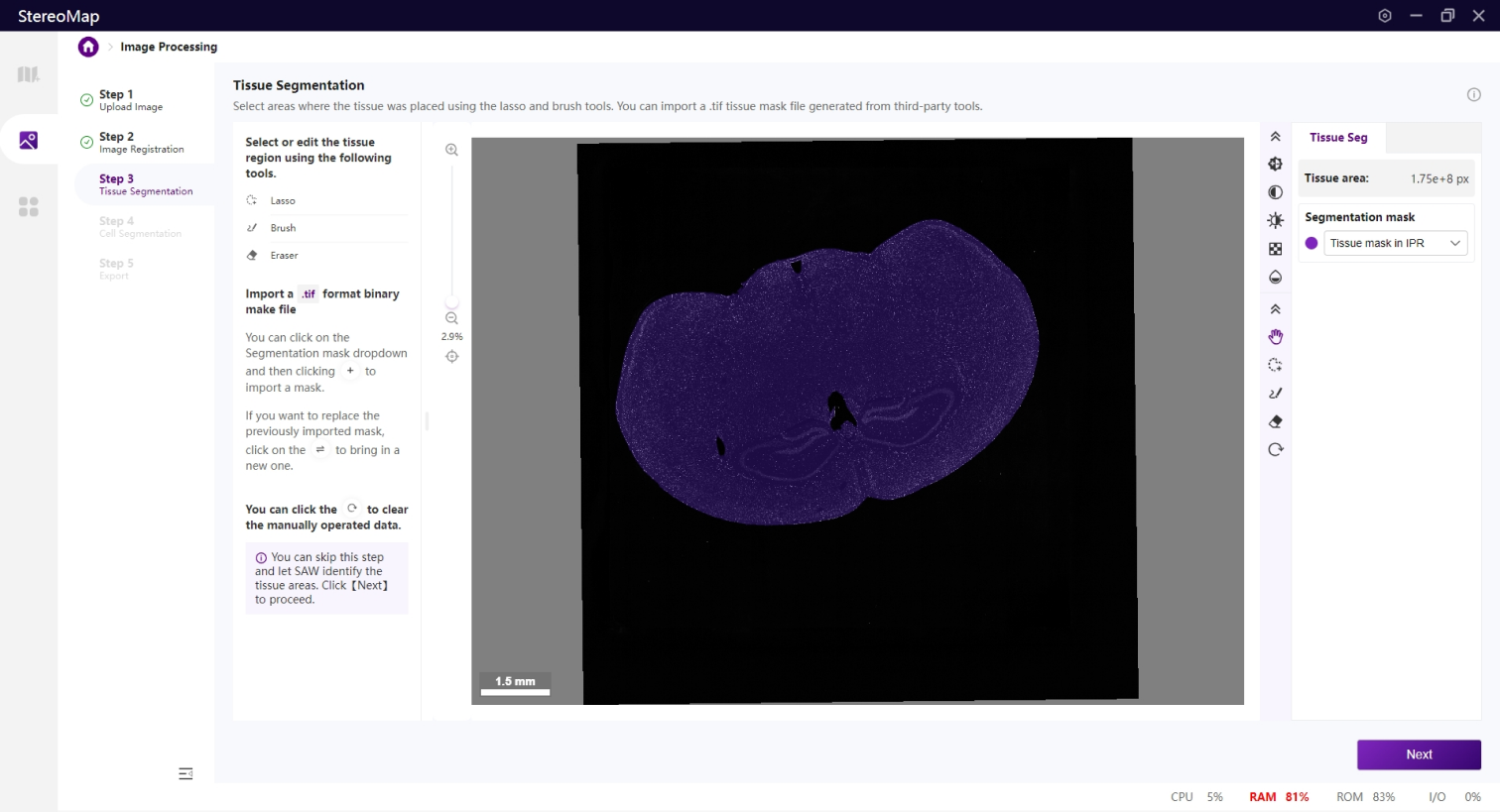
如果您在第一步中上传了.stereo文件,您可以在配准后的图像图层上看到一个半透明的组织掩膜。
如果是.tar.gz或.tif/.tiff图像文件,您需要使用手动工具来绘制组织区域。
在该步骤中,您可以编辑之前记录的组织掩膜或创建一个新的掩膜。如果是.tar.gz或.stereo文件中记录的组织掩膜将会在 Segmentation mask 下拉菜单中标记为 RECORD ,而通过手动绘制或导入创建的掩膜将标记为 CUSTOM 。如需更在画布显示的掩膜图像,只需从 Segmentation mask 下拉菜单中进行选择切换即可。
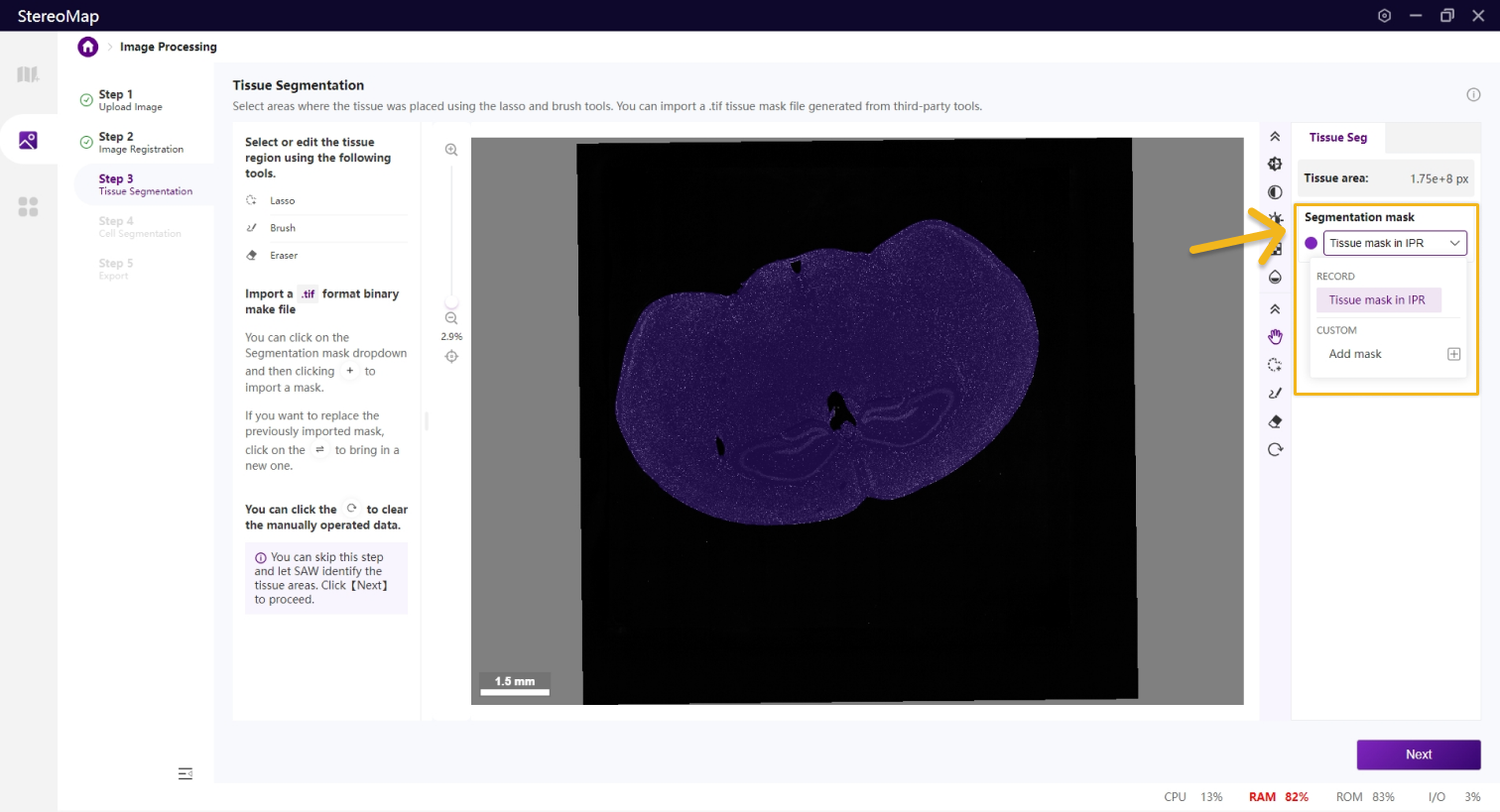
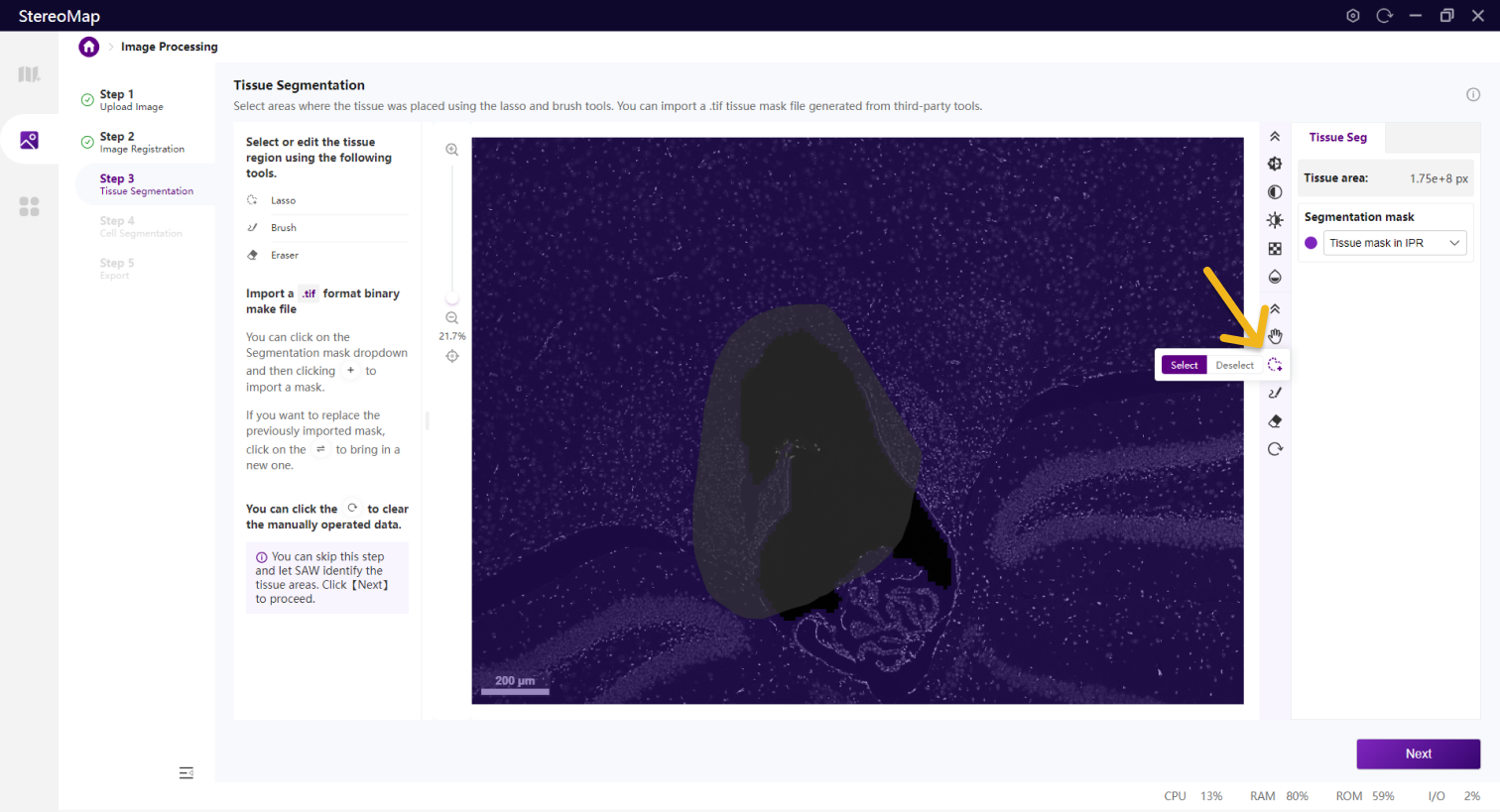
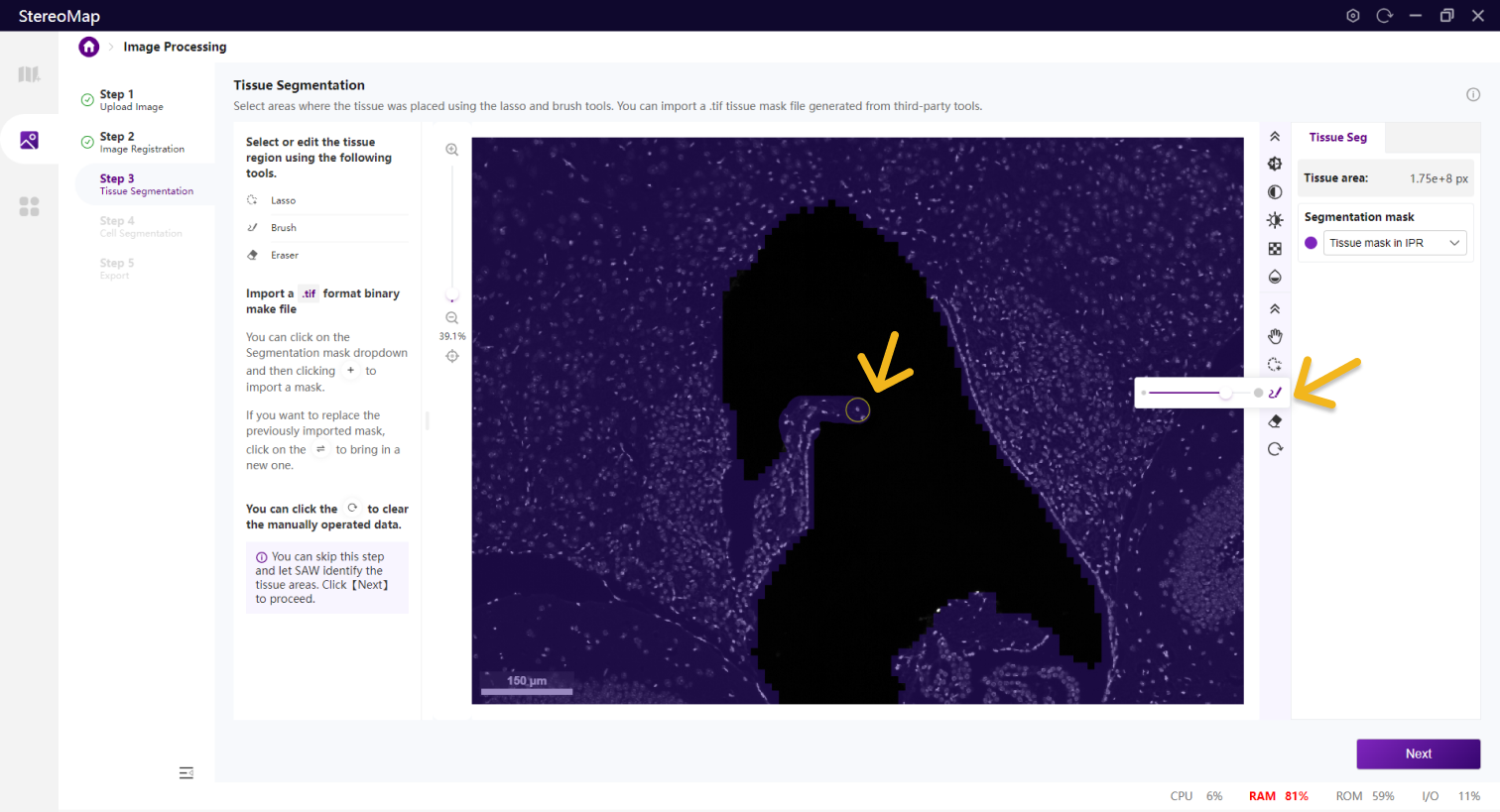
如果需要编辑组织区域的掩膜图像,可以使用套索 、画笔
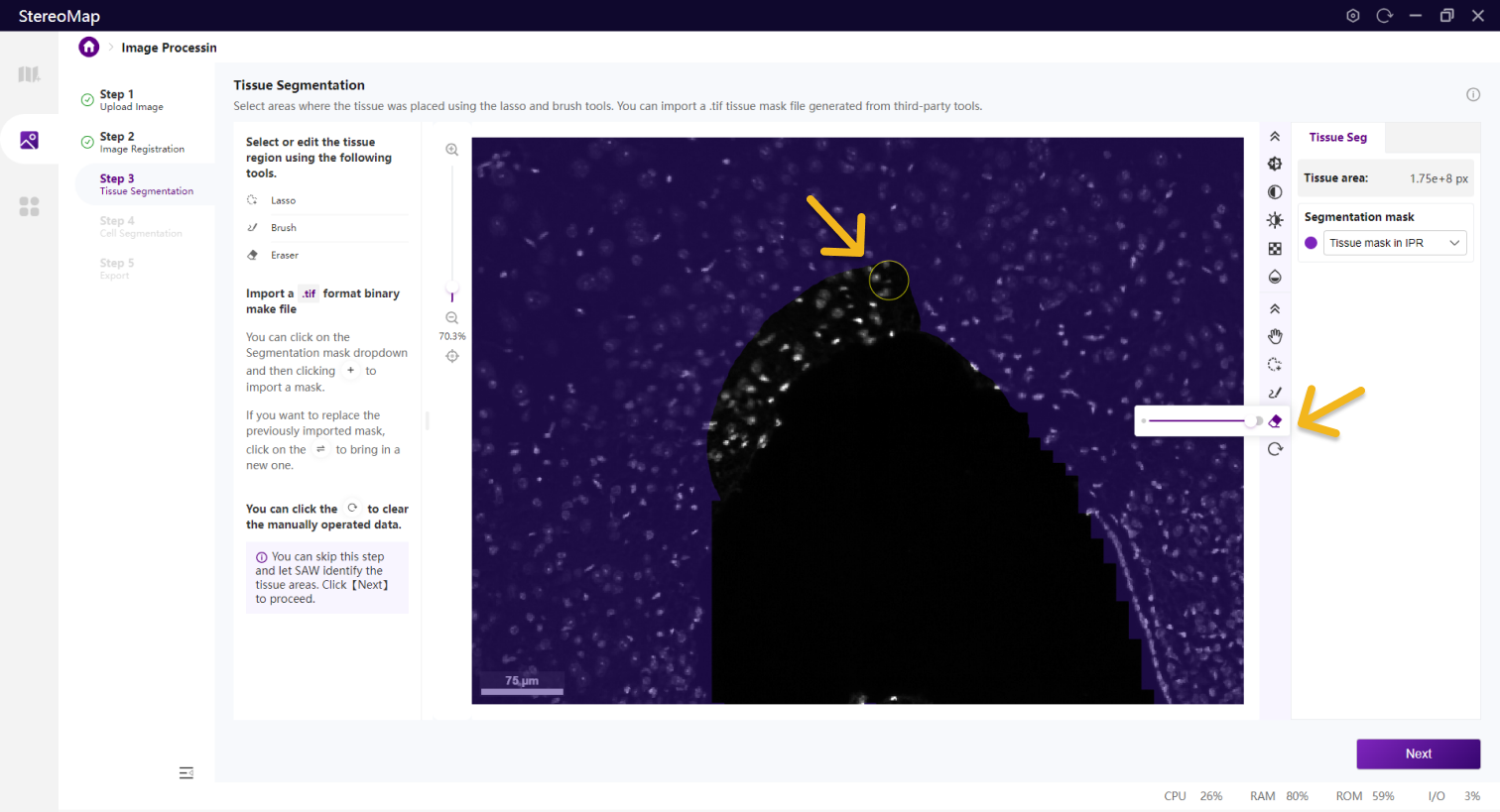
、画笔 和橡皮擦
和橡皮擦 等工具。套索一般用于选择或剔除大面积区域,而画笔和橡皮擦工具更适合于编辑组织周围或组织中的小孔等较小的区域。
等工具。套索一般用于选择或剔除大面积区域,而画笔和橡皮擦工具更适合于编辑组织周围或组织中的小孔等较小的区域。
套索
画笔
橡皮擦
您可以通过单击右侧面板上的 Segmentation mask 下拉菜单,单击来导入第三方分割工具创建的.tif格式的二进制掩膜文件,如果对于导入的结果不满意,您可以单击来替换新的掩膜文件。
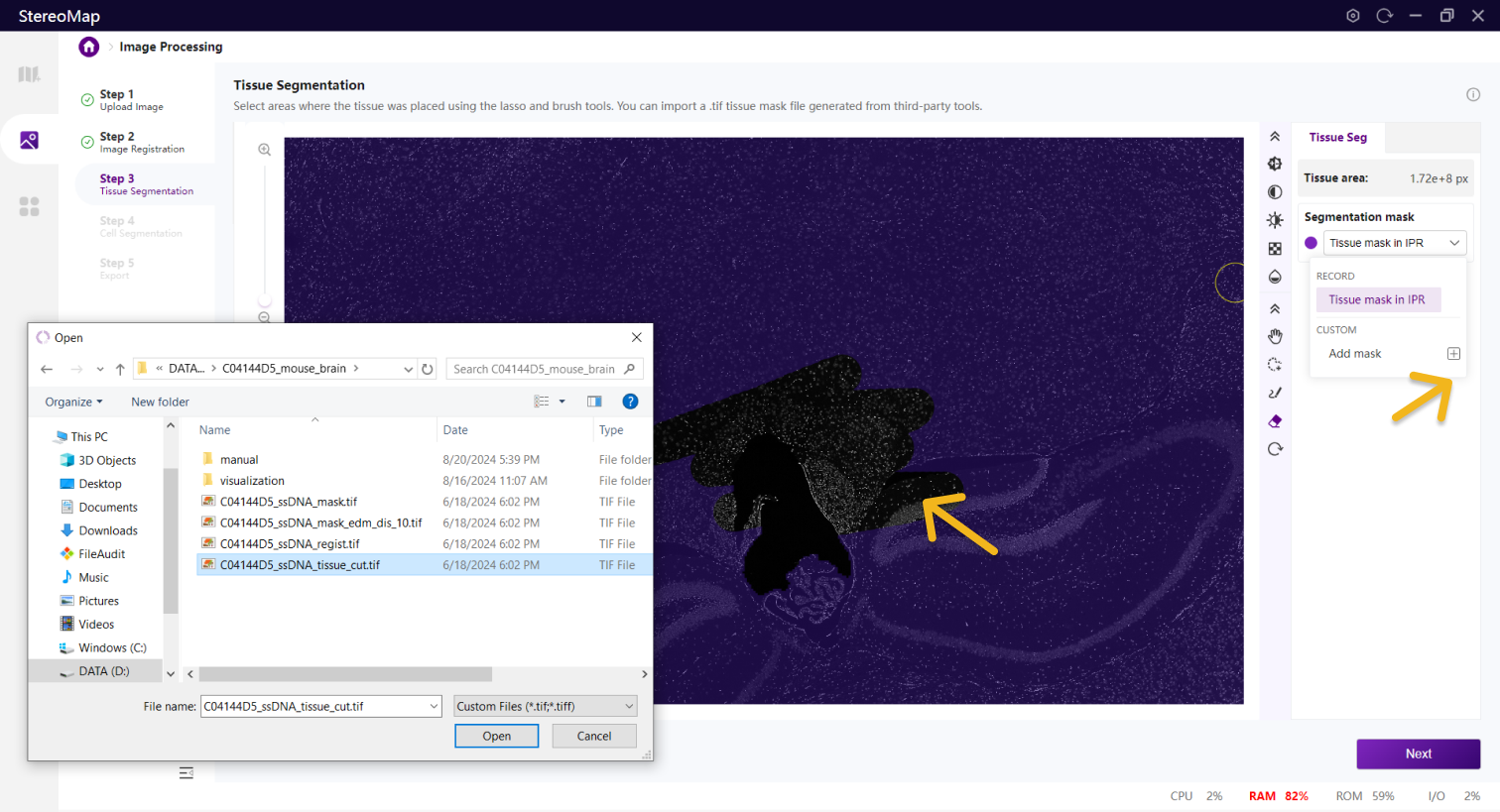
导入组织 Mask
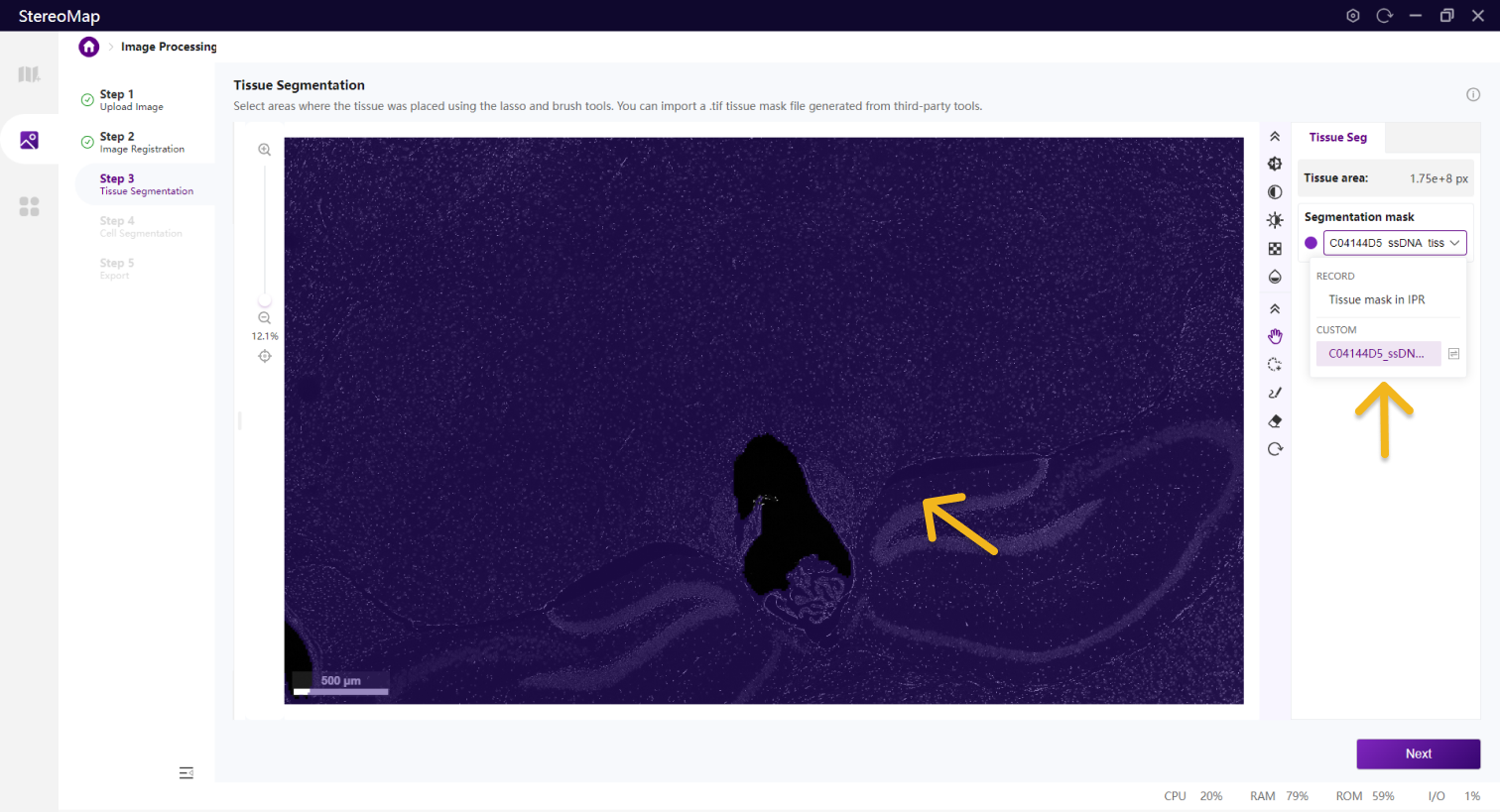
展示导入的组织 Mask 的名称
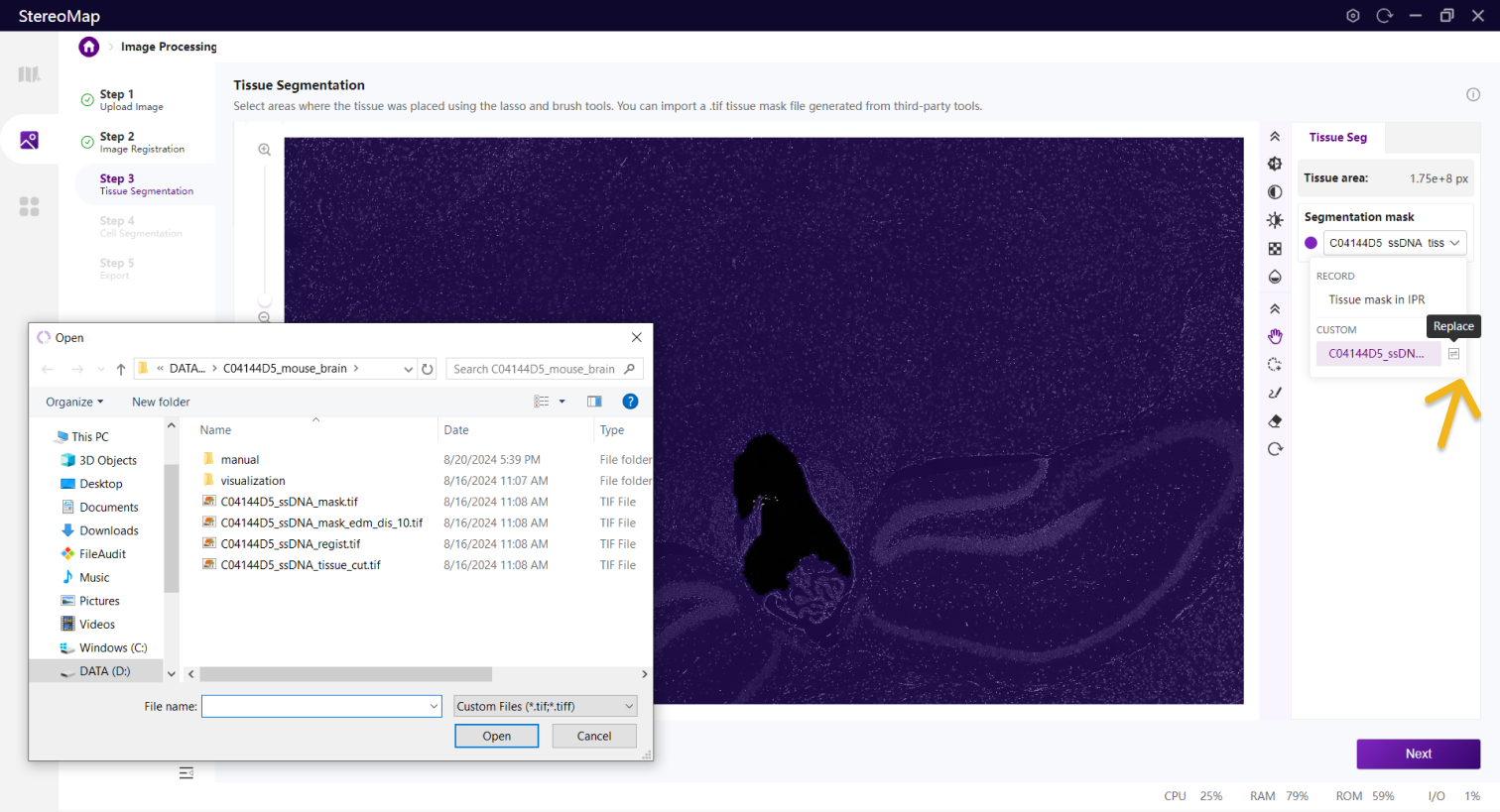
替换组织 Mask
第四步:细胞分割
细胞分割是可跳过的步骤。
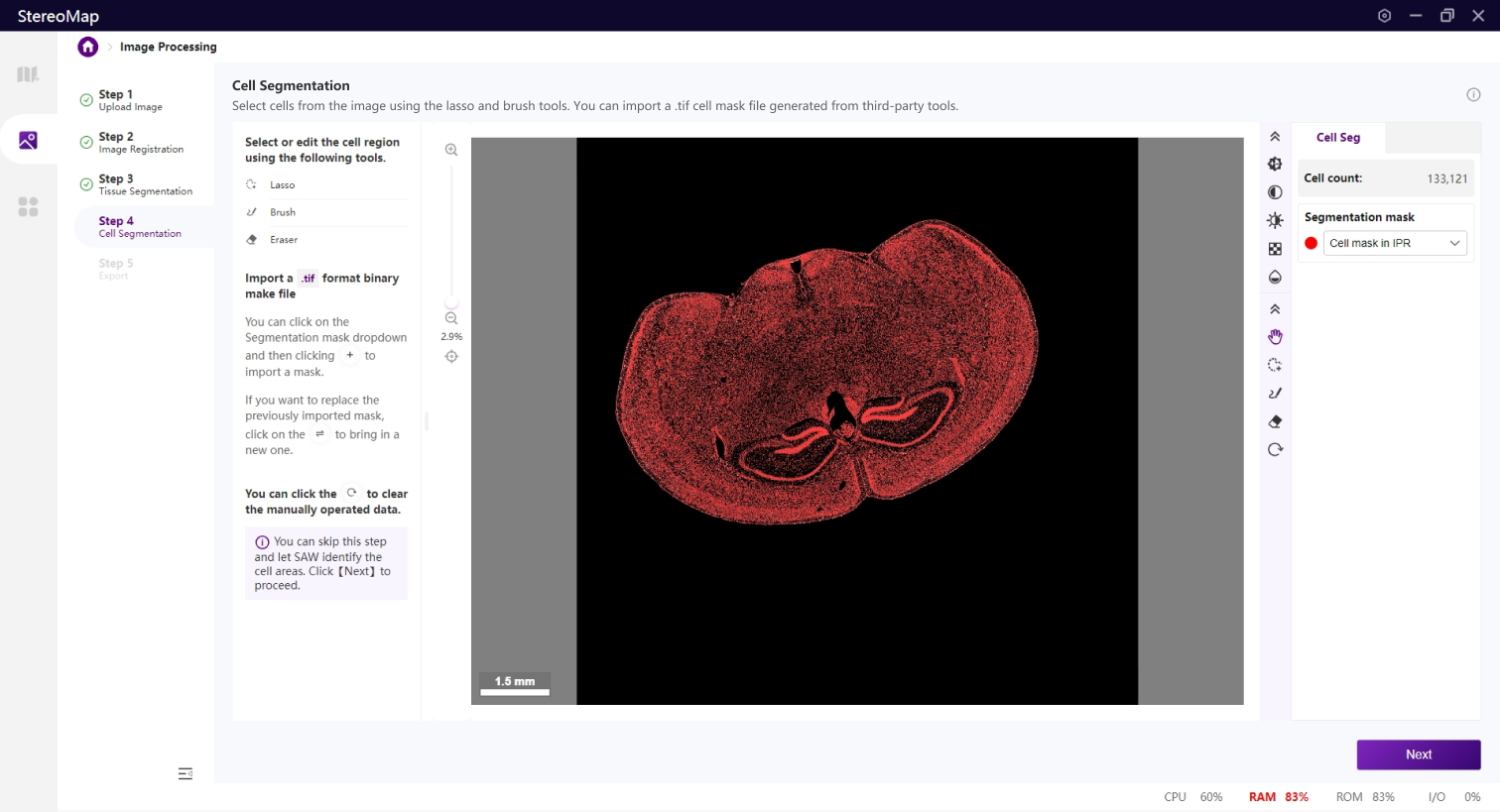
细胞分割是生成单细胞空间分辨率数据的核心步骤。
如果您在第一步中上传了.stereo文件,您可以在配准后的图像图层上看到一个红色的细胞/细胞核掩膜。
如果是.tar.gz或.tif/.tiff图像文件,您需要使用手动工具来绘制细胞。
因只在组织区域的范围内展示对应的细胞分割结果,组织外的区域将会展示为黑色的背景。
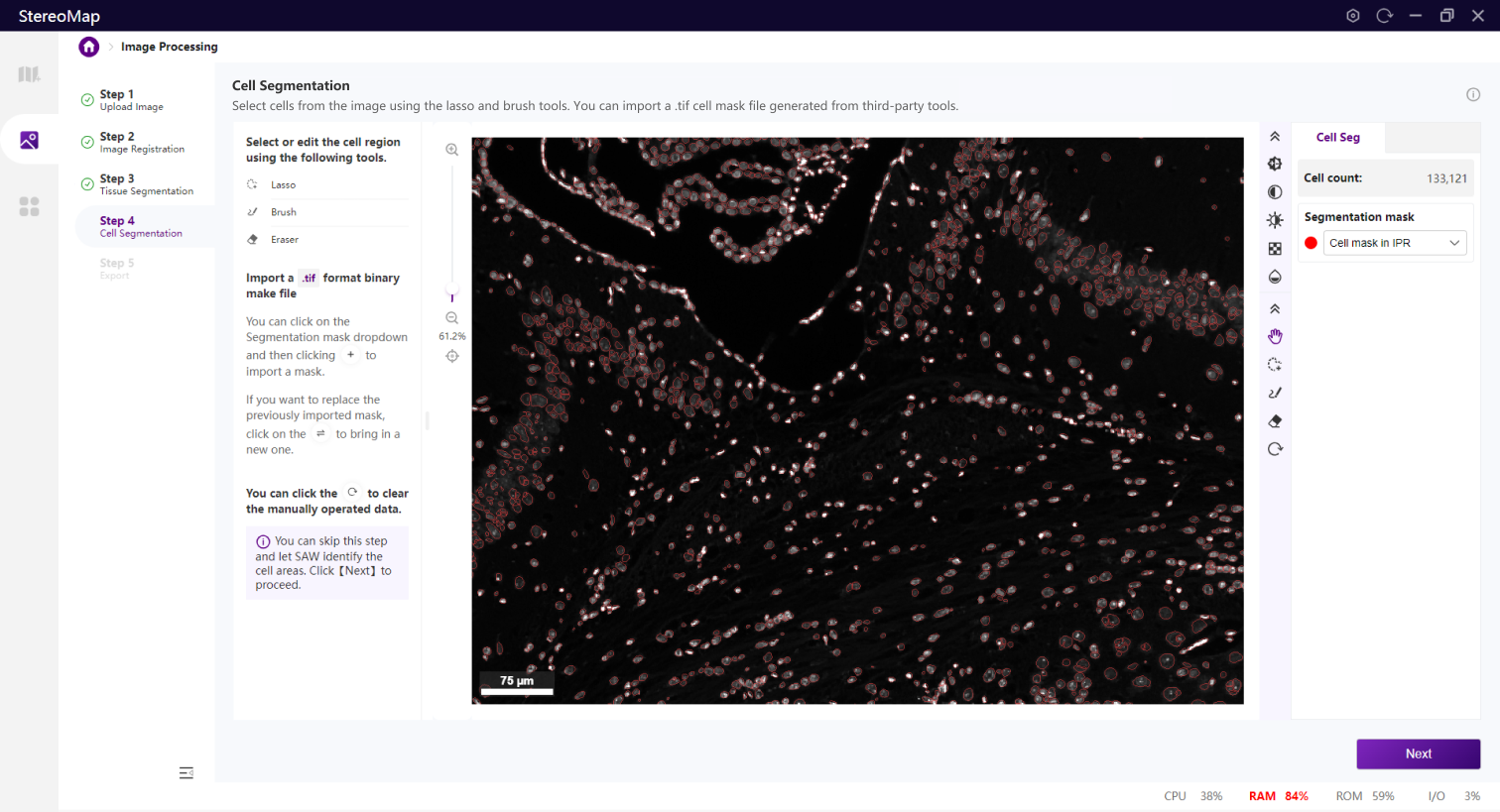
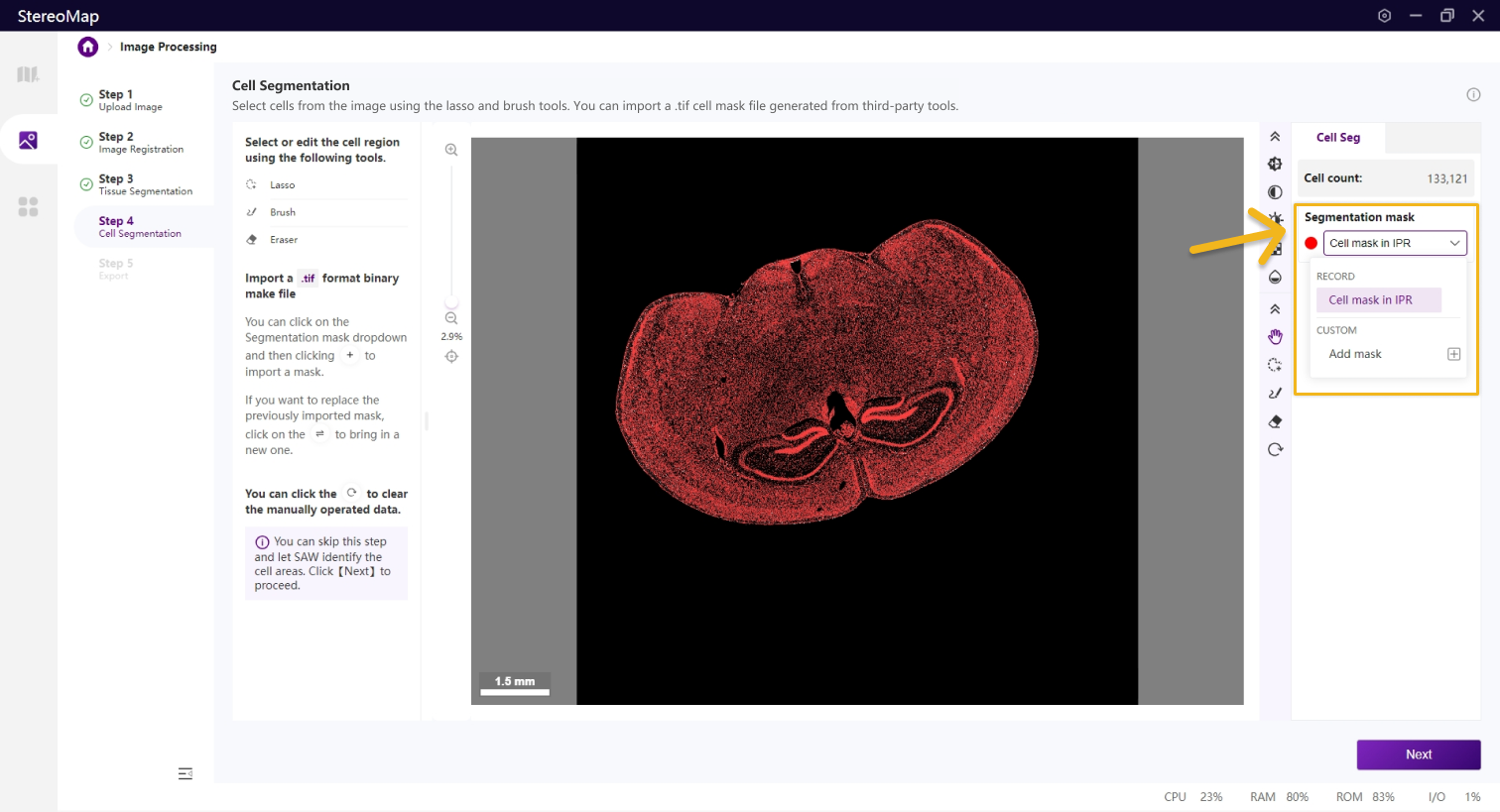
类似组织分割,您可以选择编辑之前已经记录的掩膜(标记为 RECORD )或创建一个新的掩膜(标记为 CUSTOM ),可以通过从 Segmentation mask 下拉菜单中切换画布中展示的掩膜类型。
使用套索、画笔和橡皮擦工具来编辑细胞。套索最适合用于取消大面积的背景,而画笔和橡皮擦工具更适合于较小的区域,例如标记某个细胞。
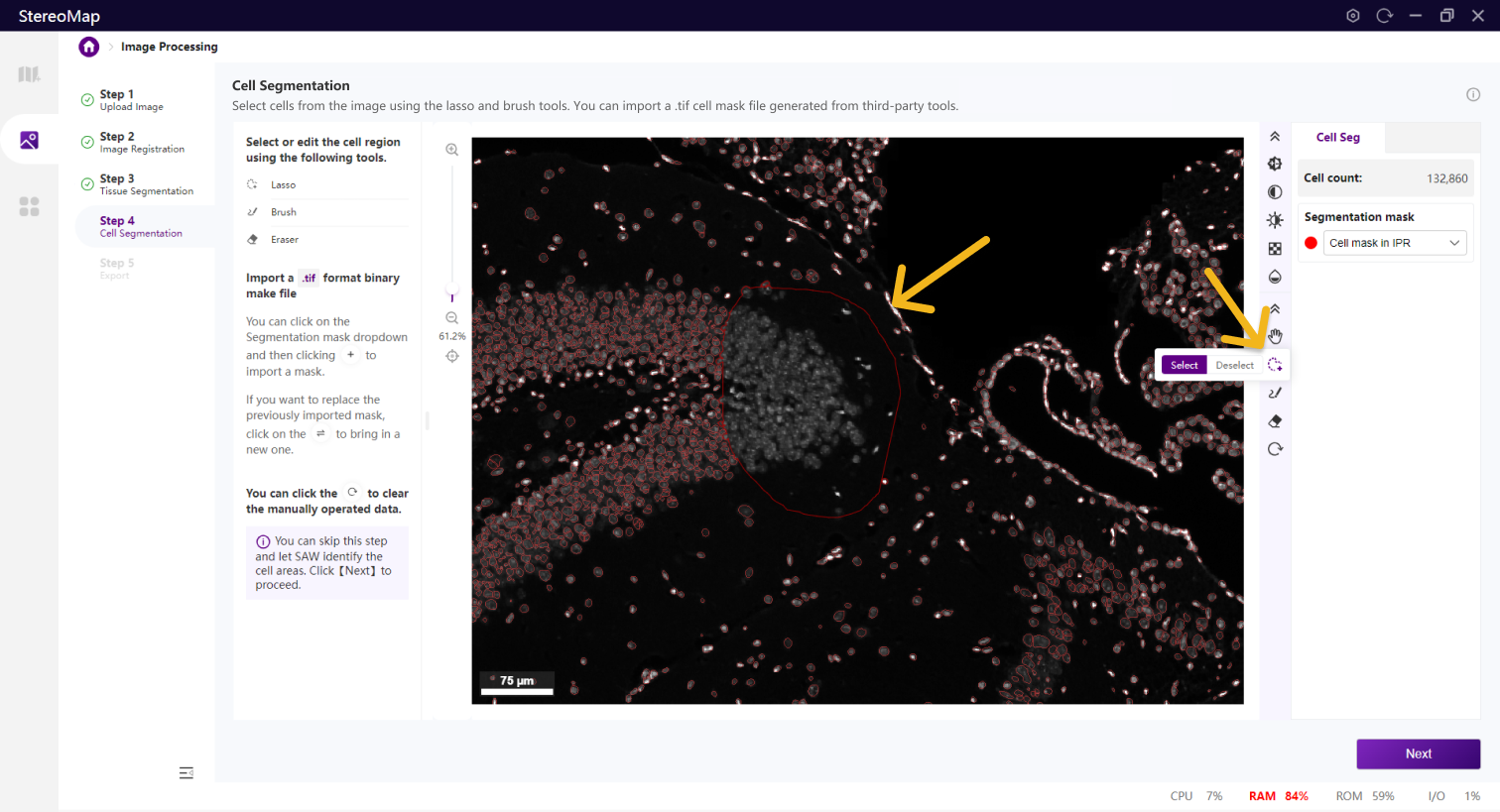
使用套索工具套索一个细胞

使用套索工具的反选删除部分细胞
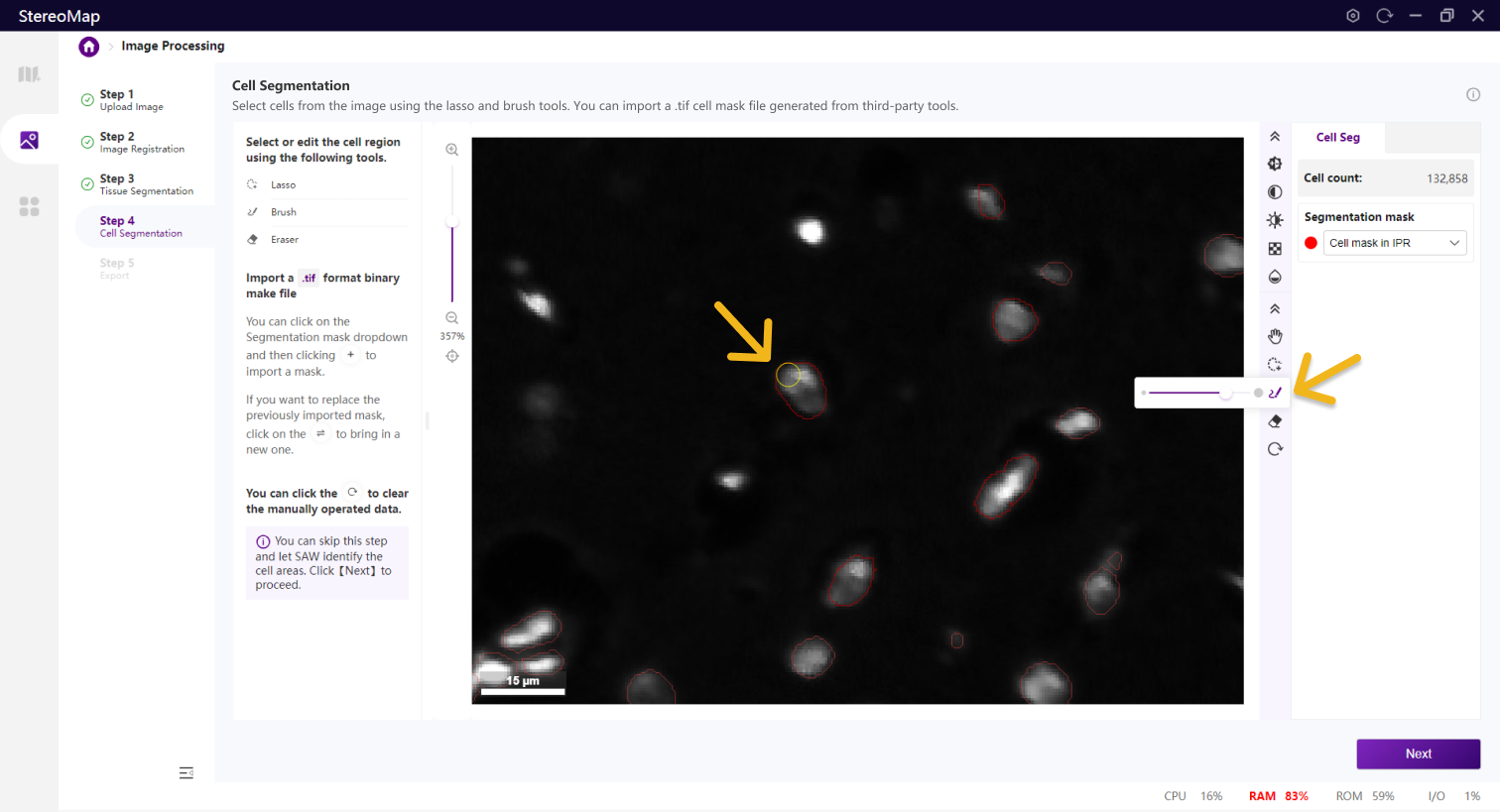
画笔
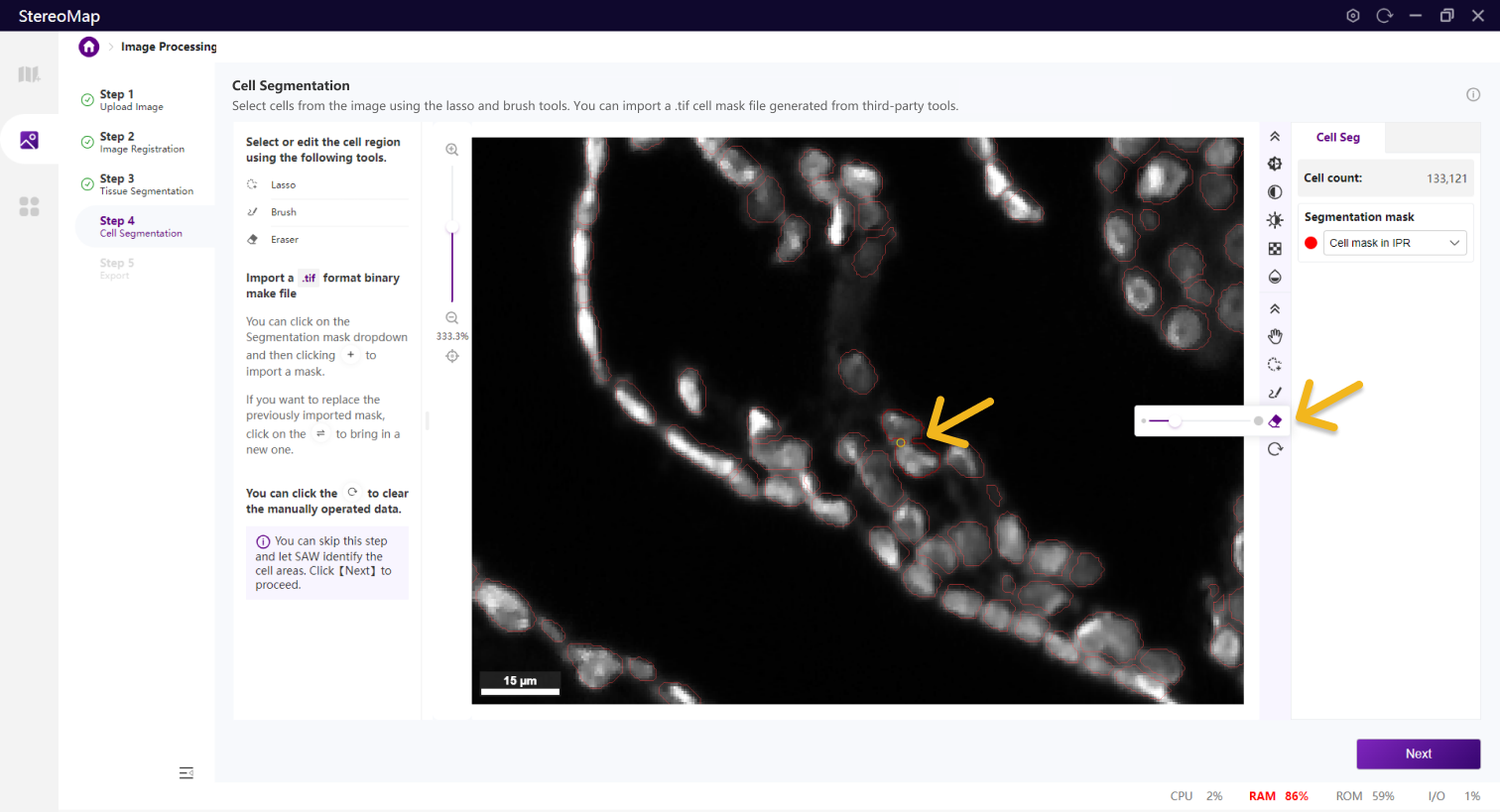
橡皮擦
建议您使用配准后的图像导入到第三方分割工具创建.tif格式的二进制细胞掩膜文件。您可以通过单击右侧面板上的 Segmentation mask 下拉菜单,单击来导入第三方分割工具分割的.tif格式的二进制掩膜文件,如果对于导入的结果不满意,您可以单击来替换新的掩膜文件。
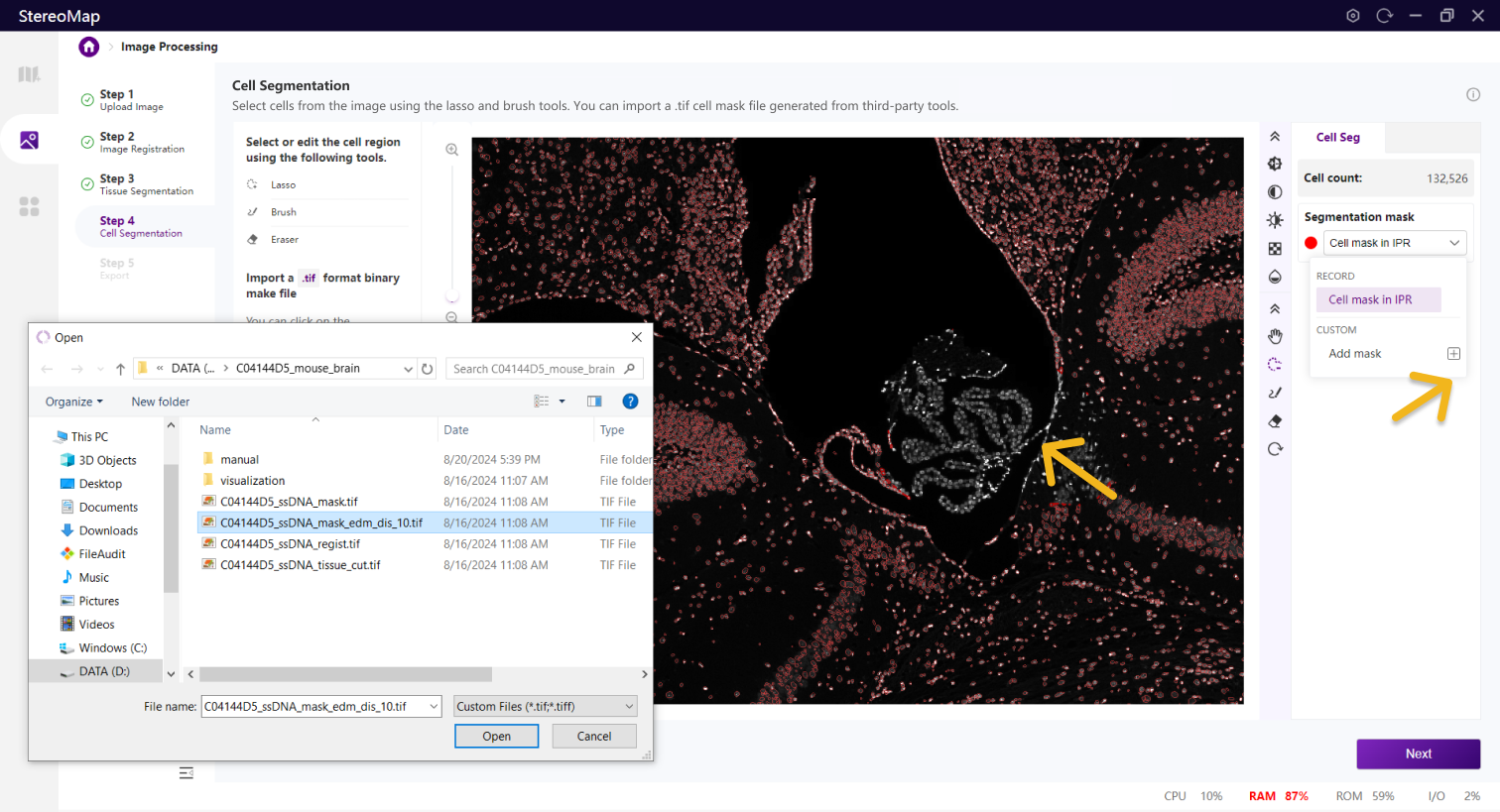
导入细胞 Mask
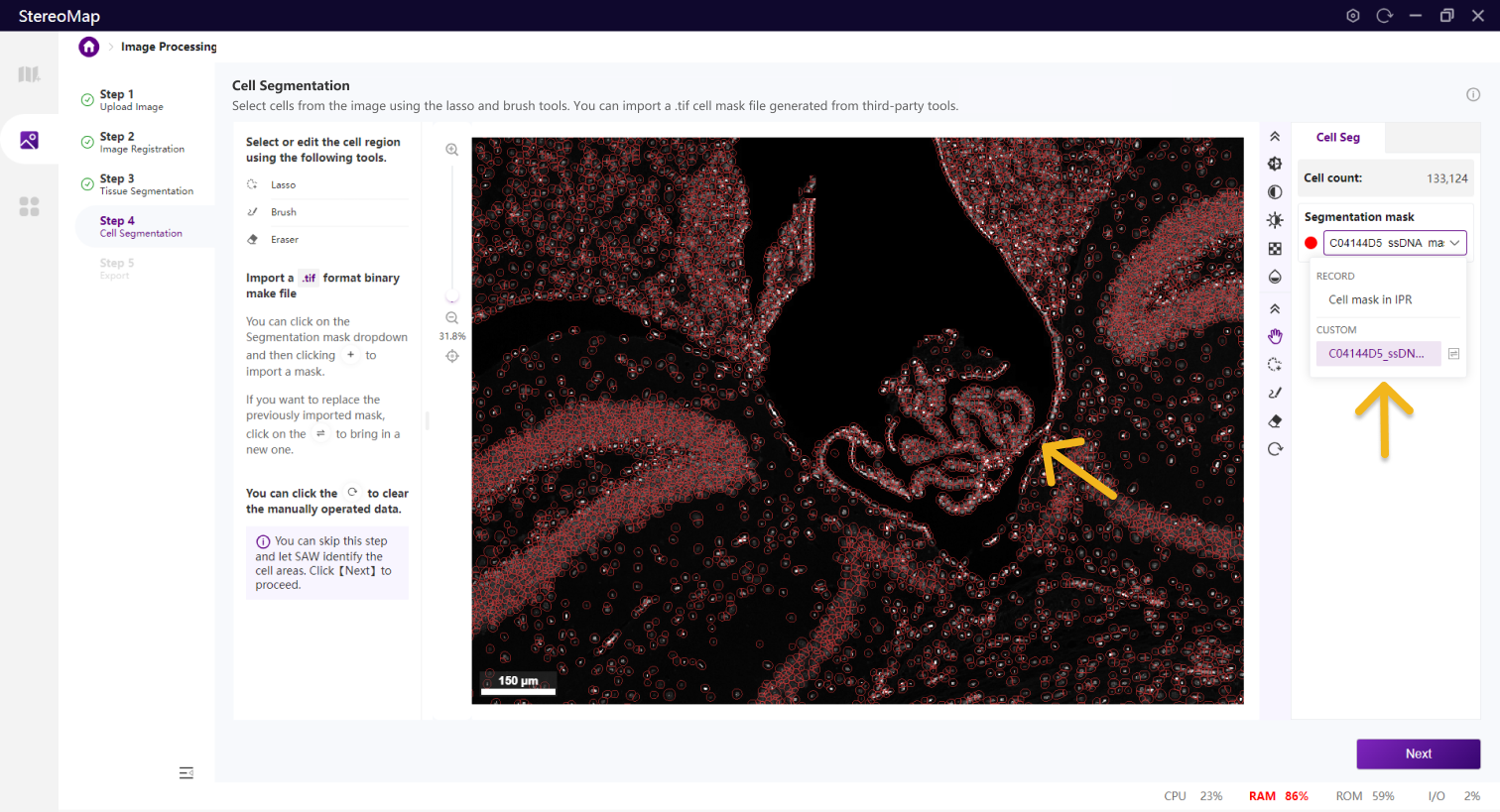
展示导入的细胞 Mask 的名称
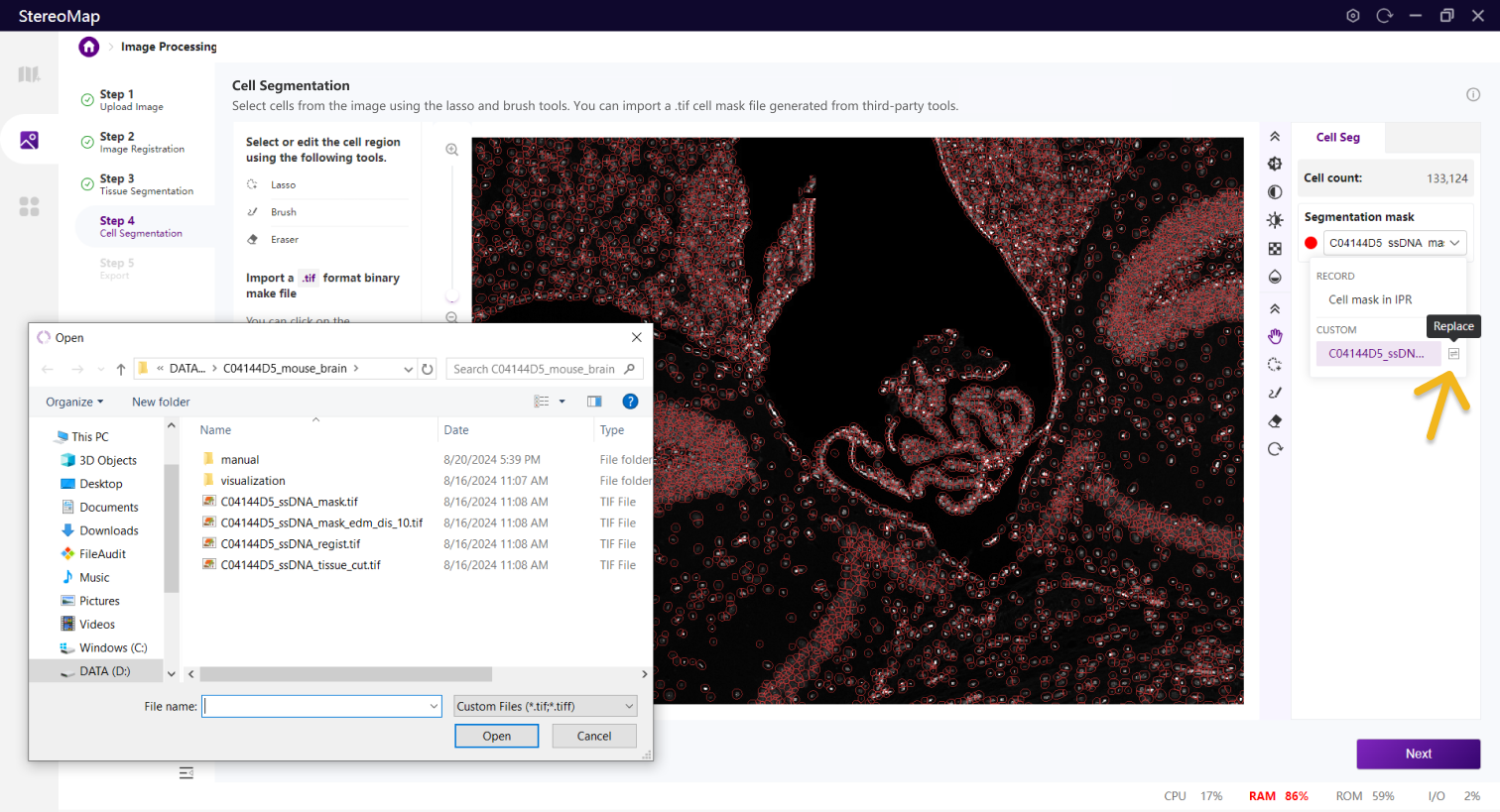
替换细胞 Mask
第五步:导出
最后是导出手动后的结果文件,单击 Export image processing record 将生成一个.tar.gz文件。
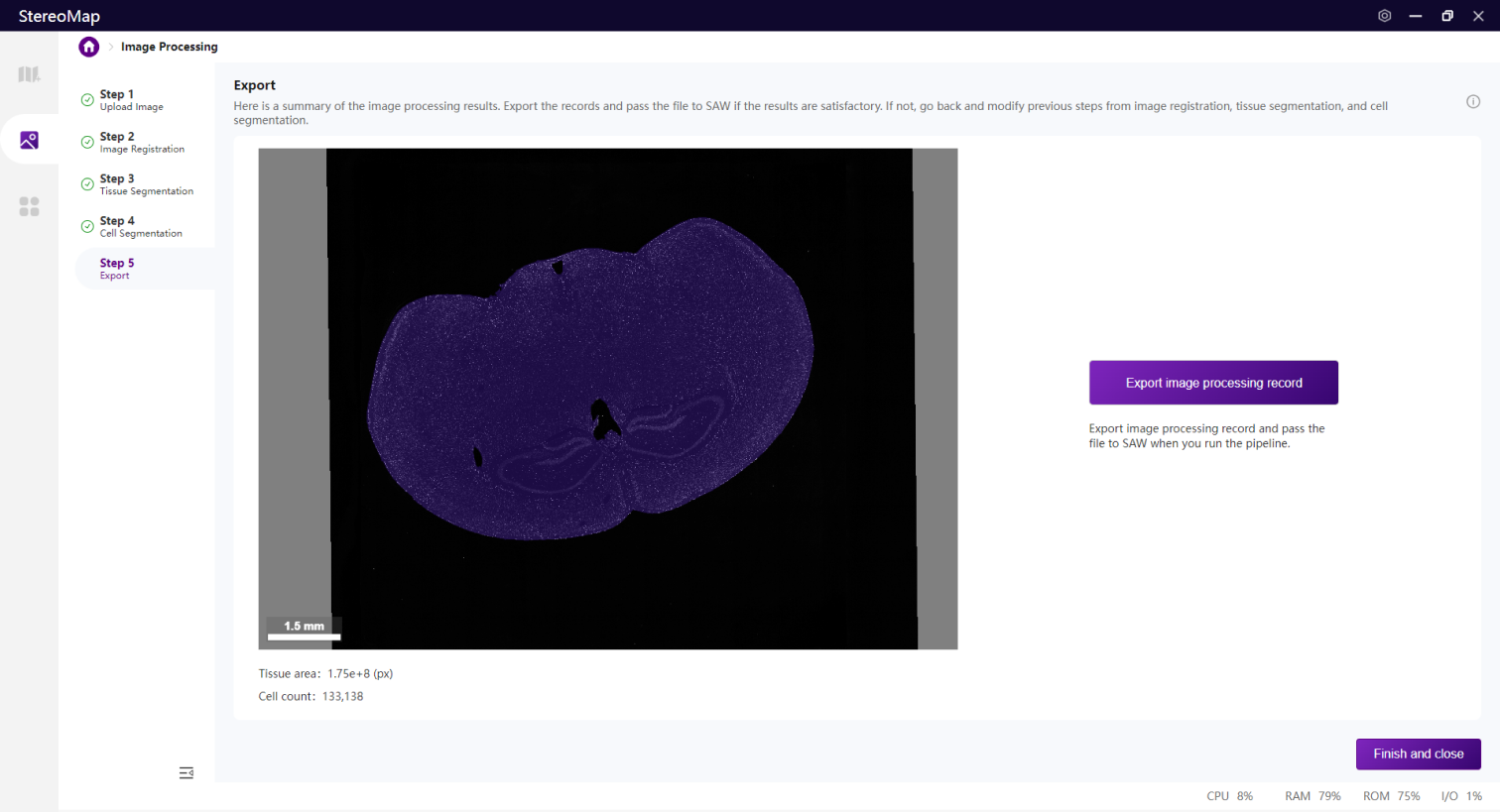
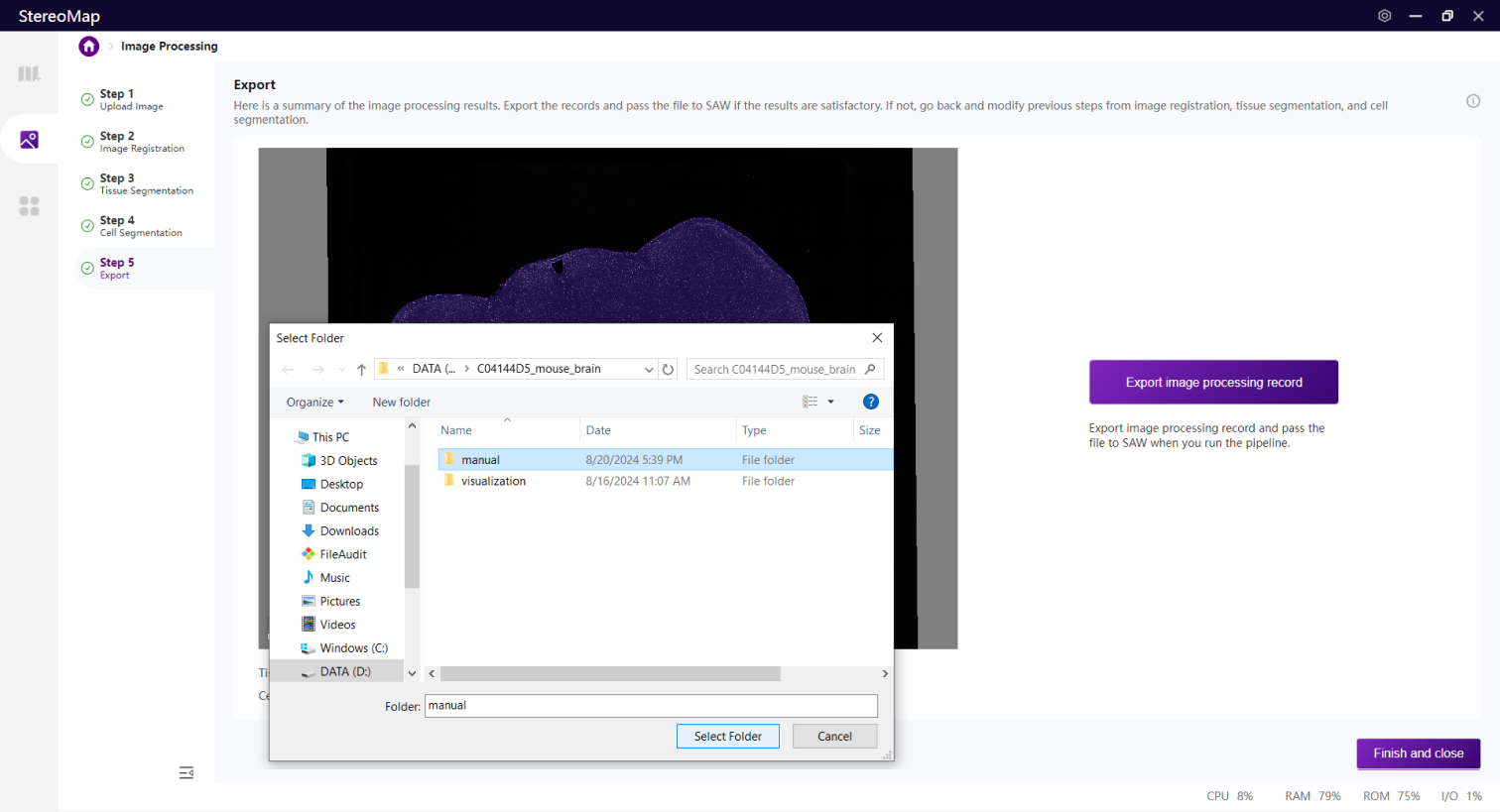
单击导出将打开您的资源管理器,选择保存的文件路径。
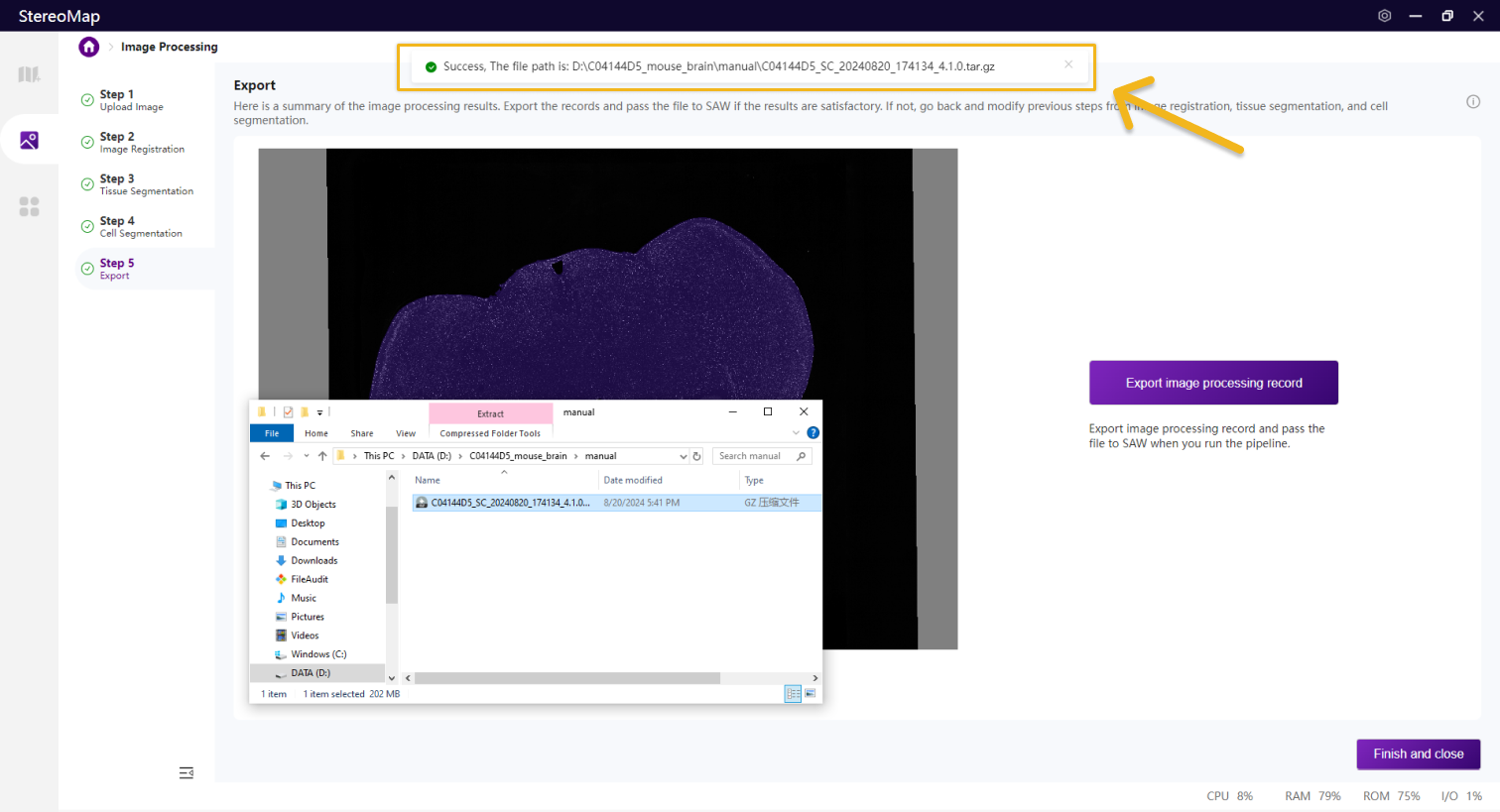
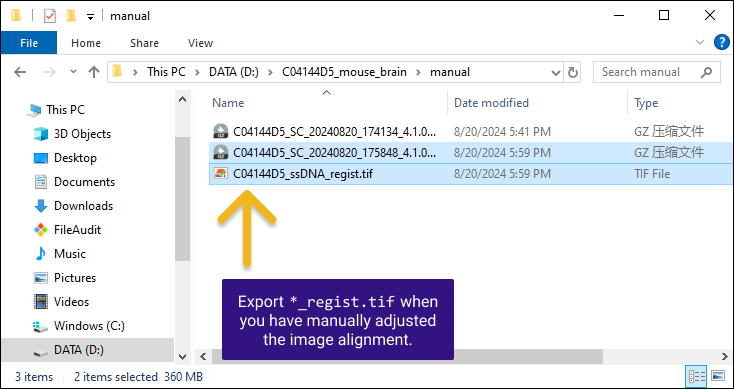
如果在第二步做了手动配准,此文件夹下会输出配准后的图像 *regist.tif ,如果您未做手动配准,该 TIFF 文件可以在 SAW 的输出目录 /outs/文件夹中找到。
.tar.gz文件包含原始的图像数据和手动处理的记录,不建议对其结构或者文件做任何修改,该文件用于在 SAW 将图像数据和测序数据一起分析。
*regist.tif文件是经过裁剪和调整的配准后的图像,跟特征表达矩阵尺寸相同。它可以在任何第三方分割工具中使用,并且生成的数据可以重新导入到 StereoMap 。
手动后的图像结果 TAR.GZ 接回 SAW
手动后的图像数据有两种选择可以接回 SAW 跑后续分析。
其中一种选择为:运行SAW count流程,将手动后的图像文件.tar.gz作为--image-tar的入参。SAW count流程将 Stereo-seq 测序的 FASTQ 文件与手动后的图像文件一起分析,并生成 HTML 报告。
cd /saw/runs
saw count \
--id=<ID> \
--sn=<SN> \
--omics=<OMICS> \
--kit-version=<TEXT> \
--sequencing-type=<TEXT> \
--chip-mask=/path/to/chip/mask \
--organism=<organism> \
--tissue=<tissue> \
--fastqs=/path/to/fastq/folders \
--reference=/path/to/reference/folder \
--image-tar=/path/to/image/tar
另一种选择为:运行SAW realign流程,将手动后的图像文件.tar.gz作为--realigned-image-tar的入参。SAW realign流程跳过比对步骤,基于手动后的结果文件重新生成配准后的图像数据,输出对应的矩阵文件,并生成 HTML 报告。
cd /saw/runs
saw realign \
--id=<ID> \
--sn=<SN> \
--count-data=/path/to/previous/SAW/count/task/folder/id \
--realigned-image-tar=/path/to/realigned/image/tar