
输出结果
SAW count 输出结果
SAW count 是处理 FASTQ 测序数据的 的核心分析流程,通常会包含显微镜图像。如果你不熟悉 SAW 分析流程的输出结果,以下内容将带你了解一些基本知识。
如果找到SAW分析的输出目录?
SAW count 分析任务通常在工作目录下执行,在该目录下,将找到一个名为 --id 或 --sn(当--id参数没有启用时)的文件夹。
输出结果下面的文件夹都是什么?
输出目录下的文件夹主要包含三部分:
/outs - 主要的分析输出结果,按照数据类型进行分类整理;
/pipeline-logs - 分析流程的日志文件和相关配置文件;
/STEREO_ANALYSIS_WORKFLOW_PROCESSING - 中间环节、临时文件,统计信息文件和单程序模块的日志文件。
主要的分析输出结果中都包含什么文件?
SAW count 分析任务运行结束后,输出的结果文件依据其数据类型被分类。主要输出文件保存在 /outs文件夹下,方便分析人员更快速找到目标文件。
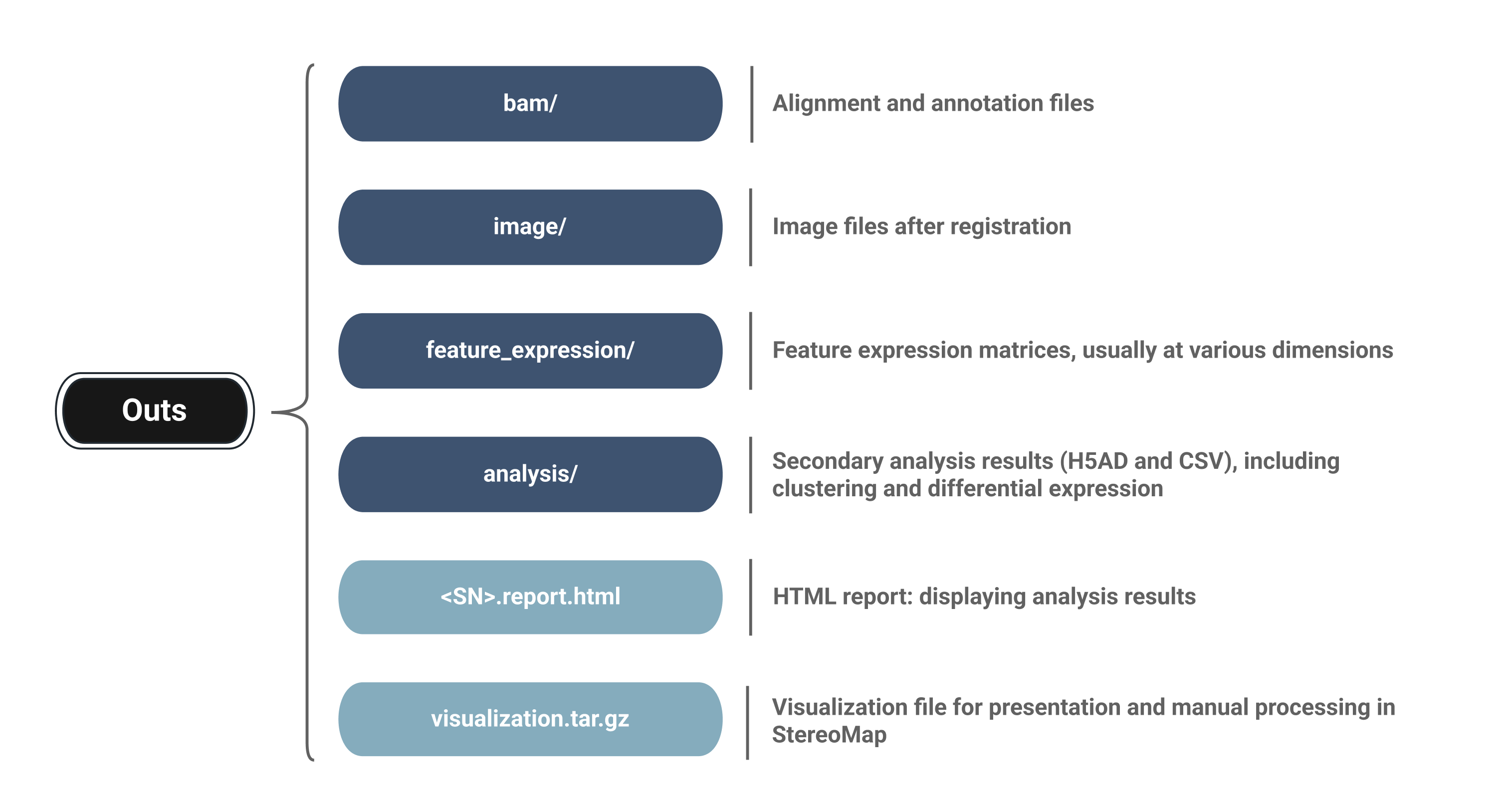
Main output files
什么是 HTML 报告?
HTML 报告汇总展示了分析过程中的统计信息,以及分析结果,以呈现 SAW count 任务运行的总体情况,更多解读请参见HTML报告详情页。
什么是可视化 visualization.tar.gz 文件?
可视化图像压缩包文件集成了 StereoMap 中结果展示所需的数据文件,其中主要包括空间表达矩阵、下游分析文件和图像相关文件。下面为一个简单的示例:
visualization
├── C04042E3.adjusted.cellbin.gef
├── C04042E3.bin20_1.0.h5ad
├── C04042E3.bin50_1.0.h5ad
├── C04042E3.cellbin_1.0.adjusted.h5ad
├── C04042E3.rpi
├── C04042E3_SC_20240930_141410_4.1.0.tar.gz
├── C04042E3.stereo
├── C04042E3.tissue.gef
└── HE_matrix_template.txt
SAW realign 输出结果
与 SAW count 的输出结果相比有何不同?
SAW realign 使用 --count-data 参数来获取reads比对、注释和空间表达矩阵相关的信息。经过手动处理的图像 .tar.gz 文件可以输出调整后的图像数据,协助分析流程生成组织和细胞维度的表达矩阵,具体结果取决于图像 .tar.gz 文件中所记录的手动处理信息。
能否通过SAW realign 分析流程输出 HTML 报告?
当然! 你可以通过输入手动处理后的图像.tar.gz文件,运行SAW realign重启分析流程。