
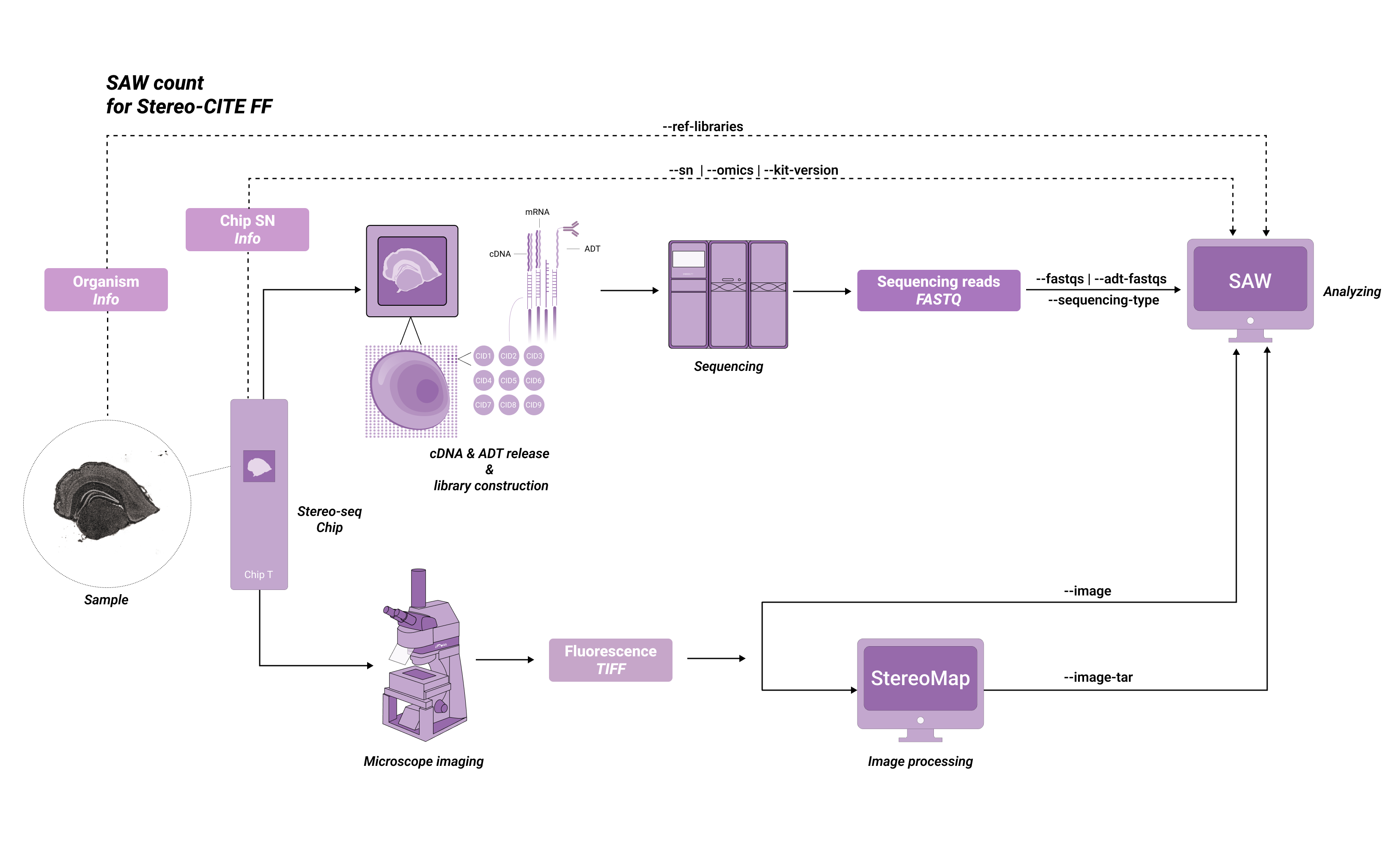
Stereo-CITE FF 分析
本教程将指导你如何使用 SAW count 进行任务分析,演示数据来自时空蛋白组转录组 Stereo-CITE 解决方案 Stereo-seq 芯片捕获的 FF (新鲜冷冻) 小鼠脾脏样本。
基本要求
为顺利运行 SAW count流程,需要先确认:
- 熟悉 Linux 系统;
- 熟悉运行命令行工具;
- 确保计算系统满足最低配置要求。
在运行分析流程之前,确保计算环境中有存储空间充足,以及运行账号有足够的权限等级。
SAW count 流程概述
使用 SAW count 分析 FF 组织样本的 Stereo-seq 测序数据。
启动分析任务前,通常需要准备好以下文件:
- 芯片 mask 文件(记录 Stereo-seq 芯片的 CID 信息)
- 基因表达和ADT (Antibody derived tag) FASTQ 测序数据(Stereo-seq 测序下机数据)
- 参考库(根据物种进行选择),包括蛋白列表和转录组参考。您可以查询 Reference libraries 帮助构建 CSV 格式的参考库。
- 一张或多张显微镜图像(
TIFF或来自 StereoMap 的图像.tar.gz)
来自 StereoMap 的压缩图像.tar.gz 文件 ,保存了原始的显微镜图像和 图像 QC 信息。
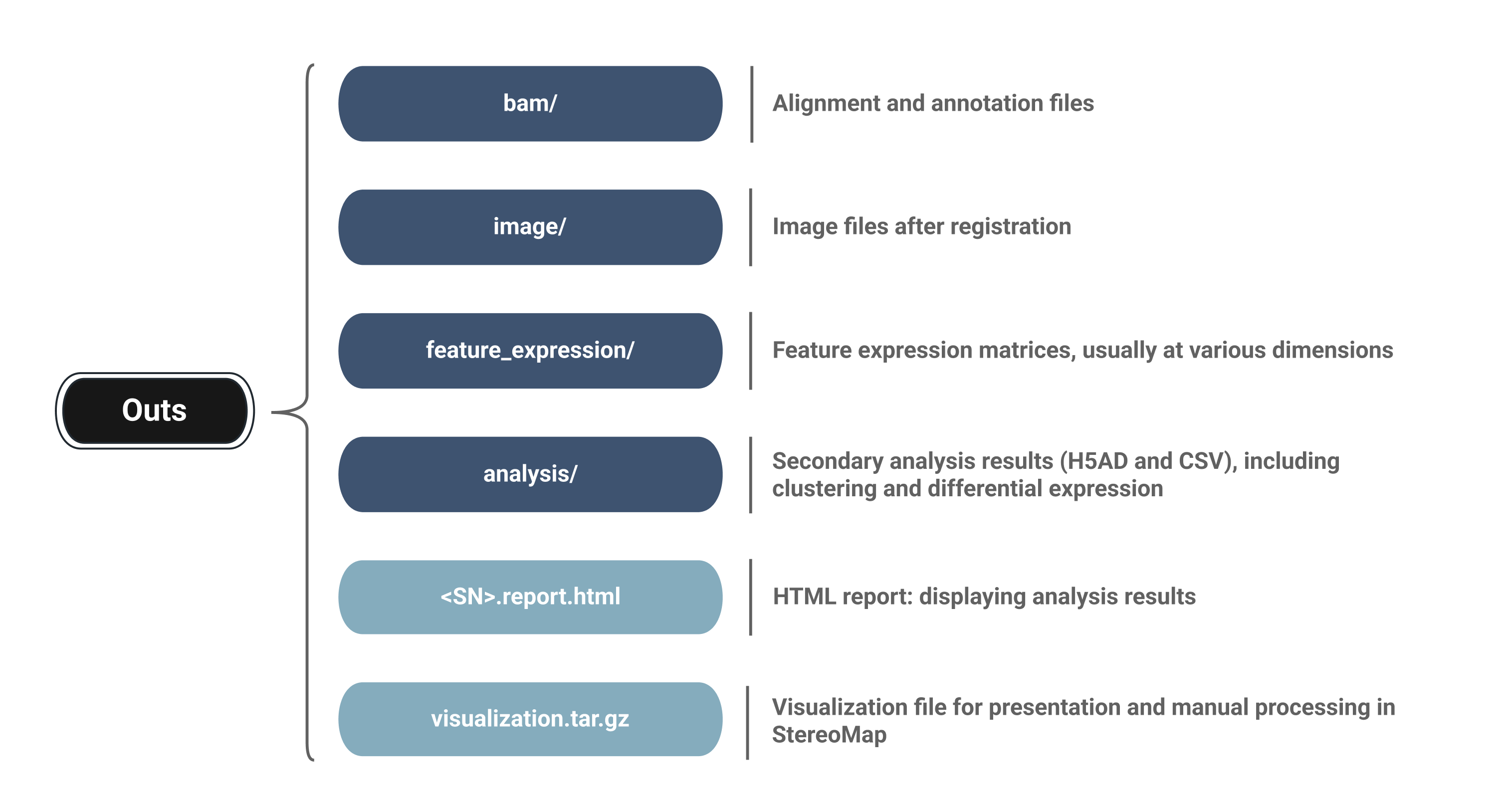
输出结果主要包括:
- 比对和注释后的 BAM 文件;
- 处理后的图像文件;
- 不同维度的基因和蛋白表达矩阵;
- 转录组聚类和差异表达分析结果;
- 蛋白组聚类结果
- 可视化文件
visualization.tar.gz, 用于 StereoMap。
Demo 数据
本教程页面使用了 Stereo-seq Chip T 捕获的小鼠脾脏组织样本。
C04776D6数据基本信息如下:
- 芯片尺寸:1cm * 1cm (S1)
- Bin1: 500 nm * 500 nm
- 10μm 厚度的组织切片
- Motic 显微镜拍摄得到的 DAPI 染色图像
在数据集页面下载芯片 mask 文件、测序 FASTQ 数据、TIFF 图像或图像 tar.gz 文件,转录组 reference 文件和蛋白列表。为了更好地归纳和整理数据,建议为不同类型的文件创建对应的文件夹。
$ cd /saw
# Create sub-folders of different datasets
$ mkdir -p datasets/STOmics-RNA-fastqs datasets/STOmics-ADT-fastqs datasets/mask datasets/image datasets/reference
参数命令
请创建参考库 ref_libraries.csv 后再提交任务。您可以查询#reference-libraries-1帮助构建参考库。
在工作目录下,运行设置 SAW count 参数命令:
saw count \
--id=<task_id> \
--sn=<SN> \
--omics=transcriptomics,proteomics \
--kit-version="Stereo-CITE T FF V1.1" \
--sequencing-type="PE75_50+100" \
--chip-mask=/path/to/chip/mask \
--organism=<organism> \
--tissue=<tissue> \
--fastqs=/saw/datasets/STOmics-RNA-fastqs \
--adt-fastqs=/saw/datasets/STOmics-ADT-fastqs \
--ref-libraries=/saw/datasets/reference/ref_libraries.csv \
--image-tar=/path/to/image/tar
命令行中参数的简要说明:
| Parameter | Description |
|---|---|
--id | (Optional, default to None) A unique task id ([a-zA-Z0-9_-]+) which will be displayed as the output folder name and the title of HTML report. If the parameter is absent, --sn will play the same role. |
--sn <SN> | (Required, default to None) SN (serial number) of the Stereo-seq chip. |
--omics <OMICS> | (Required, default to "transcriptomics") Omics information. "transcriptomics,proteomics" for Stereo-CITE analysis. |
--kit-version <TEXT> | (Required, default to None) The version of the product kit. More in count pipeline introduction. |
--sequencing-type <TEXT> | (Required, default to None) Sequencing type of FASTQs which is recorded in the sequencing report. |
--chip-mask <MASK> | (Required, default to None) Stereo-seq chip mask file. |
--organism <TEXT> | (Optional, default to None) Organism type of sample, usually referring to species. |
--tissue <TEXT> | (Optional, default to None) Physiological tissue of sample. |
--ref-libraries <CSV> | (Optional, default to None) Path to a ref_libraries.csv which declares the Transcriptome reference index built by SAW makeRef --mode=STAR , and the protein panel. Not compatible with --reference. |
--fastqs <PATH> | (Required, default to None) Path(s) to folder(s), containing all needed gene expression FASTQs. If FASTQs are stored in multiple directories, use it as: --fastqs=/path/to/directory1,/path/to/directory2,.... Notice that all FASTQ files under these directories will be loaded for analysis. |
--adt-fastqs <PATH> | (Optional, default to None) Path(s) to folder(s), containing all needed ADT FASTQs. If FASTQs are stored in multiple directories, use it as: --adt-fastqs=/path/to/directory1,/path/to/directory2,.... Notice that all FASTQ files under these directories will be loaded for analysis. |
--image <TIFF> | (Optional, default to None) TIFF image for QC (quality control), combined with expression matrix for analysis. Name rule for input TIFF : a. <SN>_<stain_type>.tifb. <SN>_<stain_type>.tiffc. <SN>_<stain_type>.TIFd. <SN>_<stain_type>.TIFF<stainType> includes: a. ssDNA b. DAPI c. HE (referring to H&E) d. <_IF_name1>_IF, <IF_name2>_IF, ... |
--image-tar <TAR> | (Optional, default to None) The compressed image .tar.gz file from StereoMap has been through prepositive QC (quality control). |
运行 SAW count
在工作目录下,运行设置 SAW count 分析任务:
cd /saw/runs
saw count \
--id=Demo_Mouse_Spleen \
--sn=C04776D6 \
--omics=transcriptomics,proteomics \
--kit-version="Stereo-CITE T FF V1.1" \
--sequencing-type="PE75_50+100" \
--chip-mask=/saw/datasets/mask/C04776D6.barcodeToPos.h5 \
--organism=mouse \
--tissue= \
--fastqs=/saw/datasets/STOmics-RNA-fastqs \
--adt-fastqs=/saw/datasets/STOmics-ADT-fastqs \
--ref-libraries=/saw/datasets/reference/ref_libraries.csv \
--image-tar=/saw/datasets/image/C04776D6_SC_20250306_110538_4.1.1.tar.gz
本教程样本的 ref_libraries.csv 内容如下
Reference,Type
/saw/datasets/reference/reference-data-mouse_SAW_v8,STAR
/saw/datasets/reference/ProteinPanel_128_mouse_V2.list,ProteinMap
如果您输入图像为 TIFF 格式,文件名的前缀应为:
<SN>_<stain_type>_*.tif
例如:
- C04144D5_ssDNA.tif
- SS200000135TL_D1_DAPI.tif
探索输出目录
分析任务运行结束后,在工作目录下会生成一个名为 Demo_Mouse_Thymus 的输出文件夹,它的命名取决于 --id 参数,当--id参数没有启用时取决于 --sn 参数的信息。
SAW count 分析任务通常在工作目录下开启,在该目录下,将找到一个名为 --id 或 --sn(当--id参数没有启用时)的文件夹。输出结果依据数据类型被分类,主要文件被保存在 /outs下。
下面列出了 SAW count 的输出文件目录结构和内容:
Demo_Mouse_Thymus
├── pipeline-logs
├── STEREO_ANALYSIS_WORKFLOW_PROCESSING
└── outs
├── analysis
├── bam
├── feature_expression
├── image
├── <SN>.report.html
└── visualization.tar.gz
进一步探究流程输出结果 :
- 跳转至HTML报告解读;
- 熟悉
visualization.tar.gz文件; - 了解输出结果中的各种文件类型