
Stereo-seq T FF
时空试剂盒信息
SAW支持分析FF (fresh frozen) 样本的时空芯片测试数据,工作流程如下图所示:
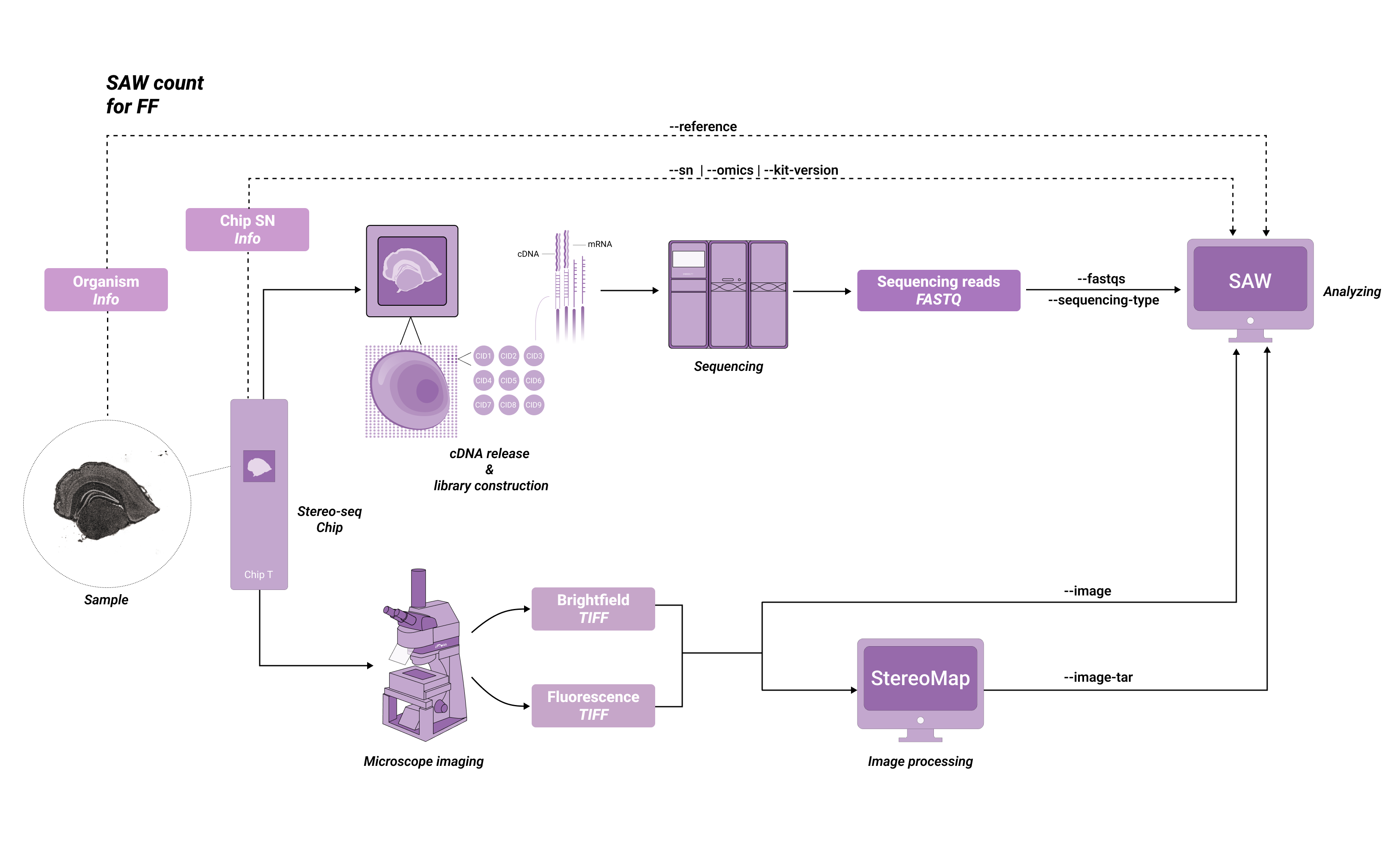
--kit-version 是开启分析前需要确认的重要信息,可参考 STOmics 的 Stereo-seq 转录组试剂套装使用说明书。
| Stereo-seq Solution | Stereo-seq Kit | --kit-version | --sequencing-type |
|---|---|---|---|
| Stereo-seq Transcriptomics Solution | V1.2.1 | "Stereo-seq T FF V1.2" | "PE100_50+100" |
| Stereo-seq Transcriptomics mIF Solution | V1.2 | "Stereo-seq T FF V1.2" | "PE100_50+100" |
| Stereo-seq Transcriptomics H&E Solution | V1.2.1 | "Stereo-seq T FF V1.2" | "PE100_50+100" |
| Stereo-seq Transcriptomics Mini Chip Solution | V1.2 | "Stereo-seq T FF V1.2" | "PE100_50+100" |
| Stereo-seq Large Chip Designs | V1.0 | "Stereo-seq T FF V1.2" | "PE100_50+100" |
| Stereo-seq Transcriptomics Solution | V1.3 | "Stereo-seq T FF V1.3" | "PE75_50+100" |
| Stereo-seq Transcriptomics mIF Solution | V1.3 | "Stereo-seq T FF V1.3" | "PE75_50+100" |
| Stereo-seq Transcriptomics H&E Solution | V1.3 | "Stereo-seq T FF V1.3" | "PE75_50+100" |
| Stereo-seq Transcriptomics Mini Chip Solution | V1.3 | "Stereo-seq T FF V1.3" | "PE75_50+100" |
| Stereo-seq Large Chip Designs | V1.3 | "Stereo-seq T FF V1.3" | "PE75_50+100" |
*T 代表 Chip T 芯片
根据上面的关系对应表,确认运行 SAW count 分析所需的 --kit-version 参数信息。
输入文件
SAW count 分析流程需要输入以下文件:
- Stereo-seq Chip T 芯片 mask 文件 (
--mask) - 时空测序 FASTQ 数据 (
--fastqs)
--sequencing-type参数信息与 FASTQ 测序数据相关,例:“PE100_50+100”,表示试剂盒采用 100 bp 双端测序方案,read 1 测序读长为 50 bp ,read 2 测序读长为 100 bp。相关信息也可以从试剂套装使用说明书中获取。
- 显微镜图像支持
TIFF或 StereoMap QC模块输出的图像.tar.gz文件--image参数支持直接输入显微镜拼接大图TIFF--image-tar参数支持输入 StereoMap QC模块输出的图像.tar.gz文件
- Reference 包含物种的参考基因组 (FASTA) 和注释文件 (GTF/GFF) (
--reference)- 参考基因组的索引文件需要预先构建,可使用
SAW makeRef - 分析流程会自动对注释文件的格式进行检查,也可以单独调用
SAW checkGTF进行格式检查
- 参考基因组的索引文件需要预先构建,可使用
运行 SAW count
根据 SAW 参数命令说明,或直接在命令行中输入 saw count --help,获取可用参数信息。在运行分析流程之前,确认已从 STOmics Cloud 平台下载芯片 mask 文件。
输入数据准备完毕后,根据图像 QC 结果选择合适的操作路径。
标准分析流程
使用 Stereo-seq FF 样本的测序数据进行基因比对和注释,输出空间特征表达矩阵,基于显微镜拍照图像,标准流程调用自动配准、组织分割、细胞分割和细胞修正算法对图像进行处理,结合表达矩阵数据和图像结果进行分析,使用下面的代码命令行开启SAW count分析:
如果是QC失败的图像数据,SAW count将不会在标准分析过程中调用图像算法,仅基于表达矩阵数据进行组织区域的识别。
cd /saw/runs
saw count \
--id=<task_id> \
--sn=<SN> \
--omics=transcriptomics \
--kit-version="Stereo-seq T FF V1.3" \
--sequencing-type="PE75_50+100" \
--chip-mask=/path/to/chip/mask \
--organism=<organism> \
--tissue=<tissue> \
--fastqs=/path/to/fastq/folders \
--reference=/path/to/reference/folder \
--image-tar=/path/to/image/tar
详细教程请参考 Stereo-seq FF 分析 部分。
图像手动处理
运行一次SAW count 标准分析流程后,可以从结果文件中获取 visualism.tar.gz 文件,下载至本地解压后,可在 StereoMap 中进行可视化查看和手动图像操作。经过一系列手动处理后,新生成的图像 .tar.gz 被传回 SAW realign,重启分析流程。
QC失败的图像数据需要在 StereoMap 中手动进行矩阵和图像之间的配准操作,之后SAW realign重启分析流程,可以基于手动配准后的结果调用图像相关的算法进行分析处理,在此过程中手动处理的其他结果(手动处理得到的组织分割和细胞分割)不会被覆盖。
cd /saw/runs
saw realign \
--id=<task_id2> \
--sn=<SN> \
--count-data=/path/to/previous/SAW/count/task/folder \
--realigned-image-tar=/path/to/realigned/image/tar
详细教程请参考 手动图像处理 部分。
输出结果
下面列出了SAW count 的输出文件目录结构和内容:
Demo_Mouse_Brain
├── pipeline-logs
├── STEREO_ANALYSIS_WORKFLOW_PROCESSING
└── outs
├── analysis
├── bam
├── feature_expression
├── image
├── <SN>.report.html
└── visualization.tar.gz
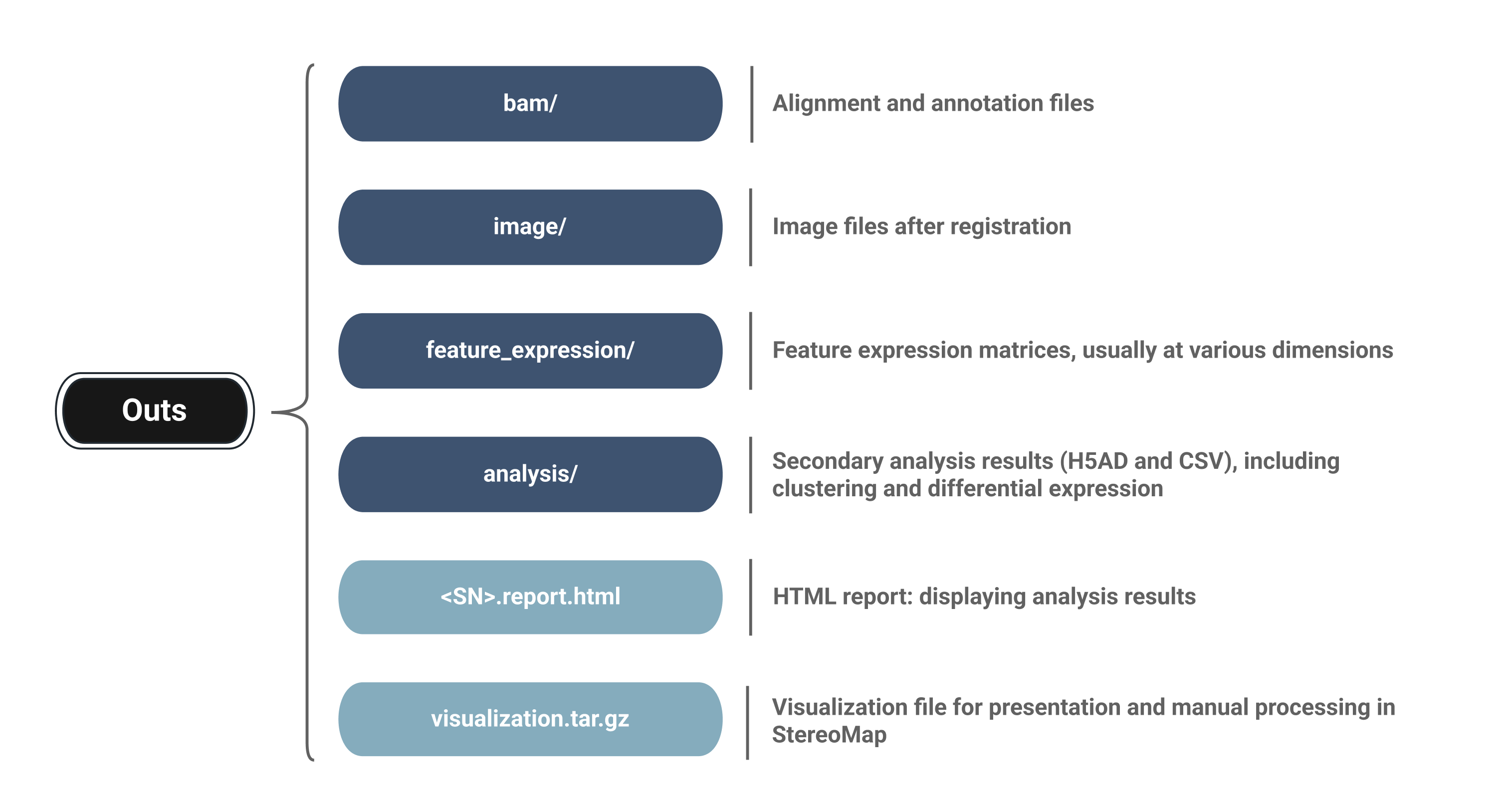
进一步探究流程输出结果 :
- 跳转至HTML报告解读;
- 熟悉
visualization.tar.gz可视化文件; - 了解输出结果中的各种文件类型。