
数据格式转换
本教程将展示如何使用辅助分析流程 SAW convert 来实现基础的文件格式转换。为了让这工具更加简易好用,SAW convert 下设置了子模块用于具体执行转换任务,子模块通常被命名为"A2B",表示从A格式转换到B格式的过程。
表达矩阵相关
visualization
转换原始 GEF 为 支持可视化的 GEF。
原始 GEF(Gene Expression File)仅在 bin1 维度上储存基因表达信息,是为了防止输出文件过大。然而,要在 StereoMap 中进行可视化,需要包含不同 bin size 的表矩阵信息,通常使用的 bin size 列表 [1, 5, 10, 20, 50, 100, 150, 200]。
saw convert visualization \
--gef=/path/to/input/GEF \
--bin-size=1,5,10,20,50,100,150,200 \
--visualization-gef=/path/to/output/visualization/GEF
gef2gem
转换 bin GEF 到 GEM。
GEM(Gene Expression Matrix)中记录的特征表达矩阵仅包含有一种 bin size,因此,在进行转换时需要设置明确的 --bin-size 参数。
saw convert gef2gem \
--gef=/path/to/input/GEF \
--bin-size=1 \
--gem=/path/to/output/GEM
转换 cellbin GEF 到 cellbin GEM。
特别注意,在转换 cellbin GEF 为 cellbin GEM 的过程中,需要获取 DNB 的信息,所以需要加入 bin GEF 文件。
saw convert gef2gem \
--cellbin-gef=/path/to/input/cellbin/GEF \
--gef=/path/to/input/bin/GEF \
--cellbin-gem=/path/to/output/cellbin/GEM
gem2gef
转换 GEM 到 bin GEF。
- 如果输入GEM 是bin1 维度的表达矩阵,则输出的GEF(基因表达文件)将是一个支持可视化的 GEF,其中包括 bin1、5、10、20、50、100、150、200 的表达矩阵信息。
- 如果输入 GEM 不是 bin1 维度的表达矩阵,输出的 GEF 将只包含特定 bin size 的表达矩阵信息。
saw convert gem2gef \
--gem=/path/to/input/GEM \
--gef=/path/to/output/GEF
转换 cellbin GEM 到 cellbin GEF。
saw convert gem2gef \
--cellbin-gem=/path/to/input/cellbin/GEM \
--cellbin-gef=/path/to/output/cellbin/GEF
bin2cell
转换 bin GEF 到 cellbin GEF。
细胞分割图记录了单个细胞的轮廓信息,可以用于提取细胞维度的表达矩阵文件。
saw convert bin2cell \
--gef=/path/to/input/GEF \
--image=/path/to/cell/segmentation/image \
--cellbin-gef=/path/to/output/cellbin/GEF \
--cellbin-gem=/path/to/output/CGEM
gef2h5ad
转换 bin GEF 到 AnnData H5AD。
AnnData H5AD 是被广泛引用于下游分析的数据格式。AnnData包版本 >= 0.8.0。
saw convert gef2h5ad \
--gef=/path/to/input/GEF \
--bin-size=20 \
--h5ad=/path/to/output/h5ad
转换 cellbin GEF 到 AnnData H5AD。
saw convert gef2h5ad \
--cellbin-gef=/path/to/input/cellbin/GEF \
--h5ad=/path/to/output/h5ad
gem2h5ad
转换 GEM 到 AnnData H5AD。
saw convert gem2h5ad \
--gem=/path/to/input/GEM \
--bin-size=20 \
--h5ad=/path/to/output/h5ad
转换cellbin GEM到AnnData h5ad。
saw convert gem2h5ad \
--cellbin-gem=/path/to/input/cellbin/GEM \
--h5ad=/path/to/output/h5ad
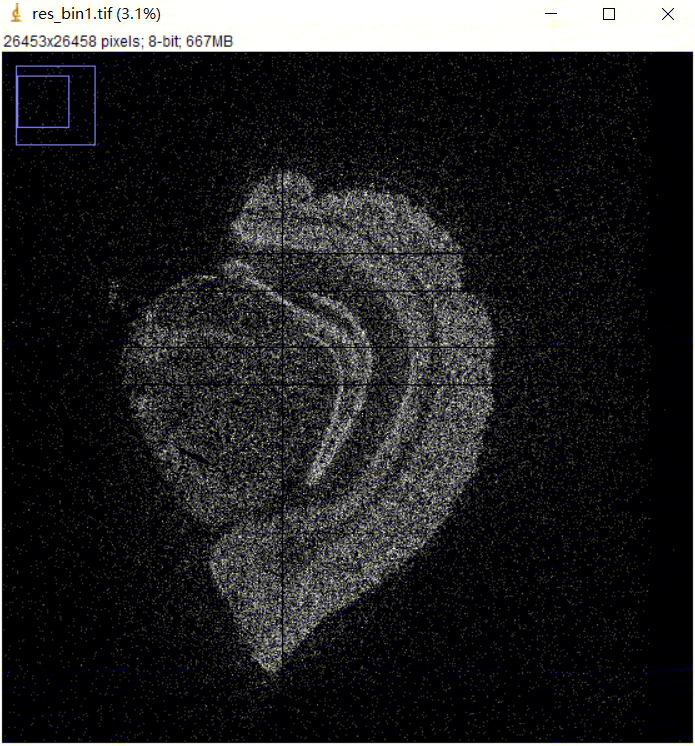
gef2img
生成 bin GEF 的灰度热图,采用灰度图展示特征表达矩阵。
saw convert gef2img \
--gef=/path/to/input/GEF \
--bin-size=1 \
--image=/path/to/output/heatmap/TIFF/image
图像相关
tar2img
从压缩图像 .tar.gz 文件中提取 TIFF 图像,其中通常包含与表达矩阵配准后的显微镜图像,组织分割图像,和细胞分割图像。自动算法或手动处理的图像结果都记录在图像 .tar.gz 文件中。
saw convert tar2img \
--image-tar=/path/to/input/image/tar \
--image=/path/to/output/folder
img2rpi
合成 TIFF 图像为 RPI 文件,用于 StereoMap 可视化展示。
Layer 图层的名字可以随意命名,但需遵从如下规则:<stain_type>/<image_type>,例如:DAPI/TissueMask。如果是细胞分割结果相关的图层,建议名称前缀为"CellMask",程序会自动将其转换为细胞轮廓进行展示。
saw convert img2rpi \
--image=/path/to/input/image1,/path/to/input/image2,/path/to/input/image3... \
--layers=DAPI/Image,DAPI/TissueMask,DAPI/CellMask... \
--rpi=/path/to/output/rpi
merge
合并多张图为一张图像(至多3张图)。
请注意,图像输入的顺序代表其颜色通道 R-G-B。
saw convert merge \
--image=/path/to/input/image1,/path/to/input/image2,/path/to/input/image3 \
--merged-image=/path/to/output/multichannel/image
合并显微镜图像 ssDNA_SS200000135TL_D1_regist.tif 和组织分割图 ssDNA_SS200000135TL_D1_tissue_cut.tif,可用于评估组织分割的效果。

显微镜图像 ssDNA_SS200000135TL_D1_regist.tif 和细胞分割图 ssDNA_SS200000135TL_D1_mask.tif 合并结果的部分区域,用于评估细胞分割的效果。
.png)
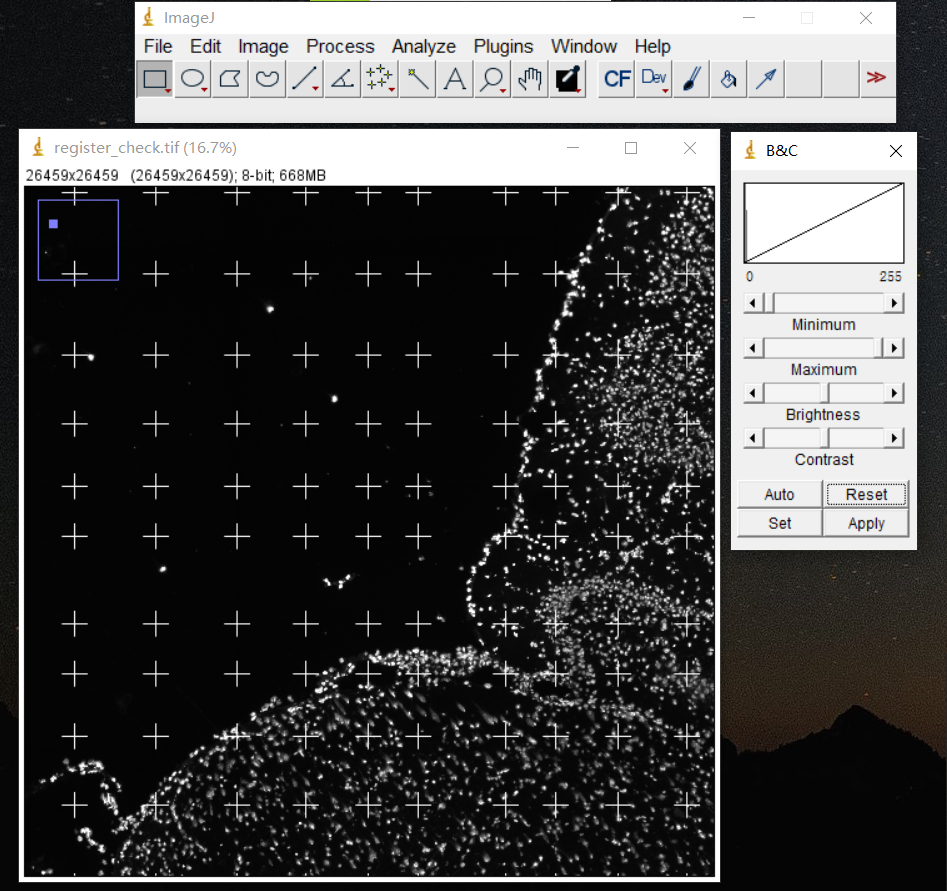
overlay
将通过矩阵推导的模板点叠加到图像上,用以检查图像 QC 识别到的图像模板点是否准确。
矩阵模板文件 <stain_type>_matrix_template.txt 可在 可视化文件 visualization.tar.gz 中找到。
saw convert overlay \
--image=/path/to/input/image \
--template=/path/to/input/template/txt \
--overlaid-image=/path/to/output/overlaid/image
将矩阵模板点叠加到 ssDNA_SS200000135TL_D1_regist.tif 图像上,用以检查配准效果。
.png)